Structural and Computational Biology Unit, European Molecular Biology Laboratory, Heidelberg, Germany.
Bioessays. 2011 Oct;33(10):769-80. doi: 10.1002/bies.201100062. Epub 2011 Aug 19.
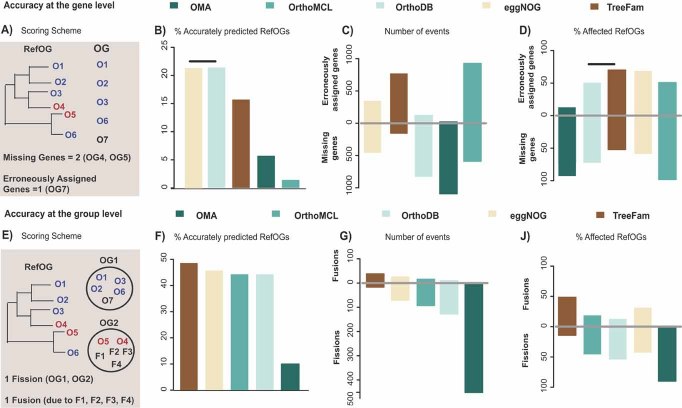
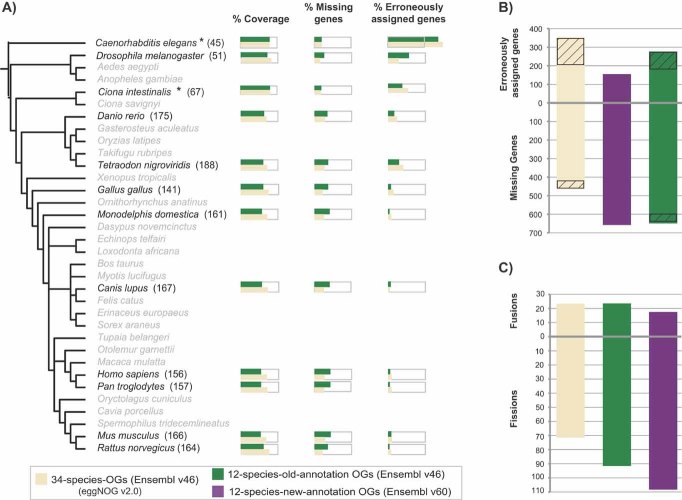
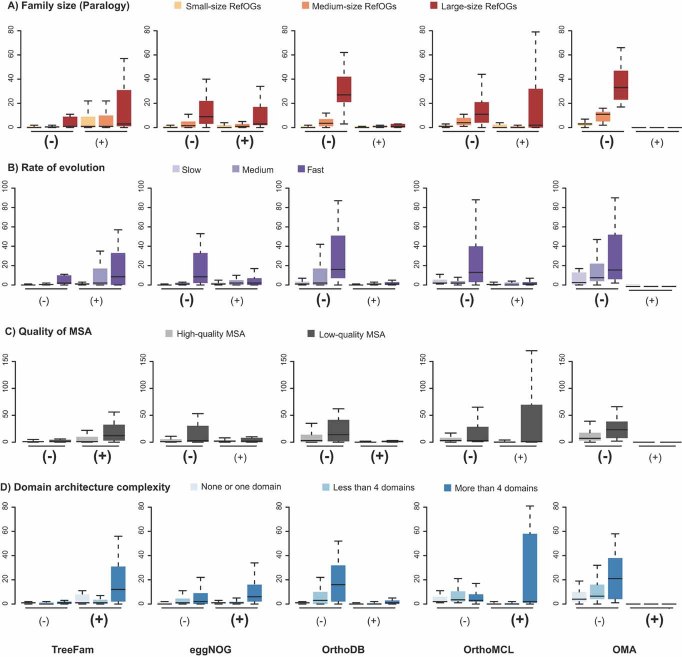
The increasing number of sequenced genomes has prompted the development of several automated orthology prediction methods. Tests to evaluate the accuracy of predictions and to explore biases caused by biological and technical factors are therefore required. We used 70 manually curated families to analyze the performance of five public methods in Metazoa. We analyzed the strengths and weaknesses of the methods and quantified the impact of biological and technical challenges. From the latter part of the analysis, genome annotation emerged as the largest single influencer, affecting up to 30% of the performance. Generally, most methods did well in assigning orthologous group but they failed to assign the exact number of genes for half of the groups. The publicly available benchmark set (http://eggnog.embl.de/orthobench/) should facilitate the improvement of current orthology assignment protocols, which is of utmost importance for many fields of biology and should be tackled by a broad scientific community.
随着测序基因组数量的不断增加,已经开发出了几种自动化的直系同源预测方法。因此,需要进行测试以评估预测的准确性,并探索由生物学和技术因素引起的偏差。我们使用了 70 个经过人工整理的家族来分析 Metazoa 中五种公共方法的性能。我们分析了方法的优缺点,并量化了生物学和技术挑战的影响。从分析的后半部分可以看出,基因组注释成为最大的单一影响因素,最多可达 30%的性能受到影响。通常,大多数方法在分配直系同源群方面表现良好,但它们未能为一半的群组分配确切的基因数量。公开可用的基准集(http://eggnog.embl.de/orthobench/)应该有助于改进当前的直系同源分配协议,这对生物学的许多领域都至关重要,应该由广泛的科学界来解决。