Centre for Computational Chemistry, School of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, United Kingdom.
J Am Chem Soc. 2011 Oct 5;133(39):15464-74. doi: 10.1021/ja203157u. Epub 2011 Sep 12.
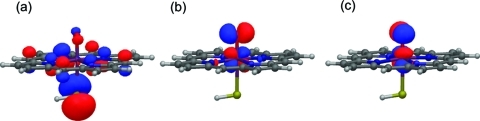
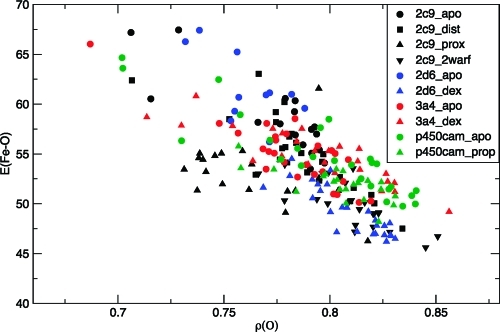
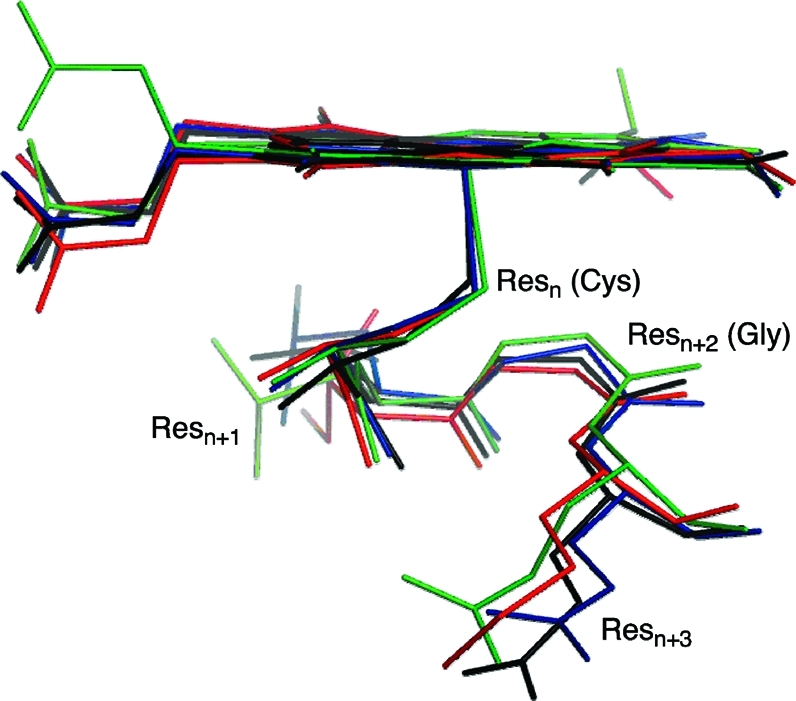
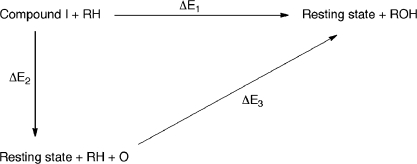
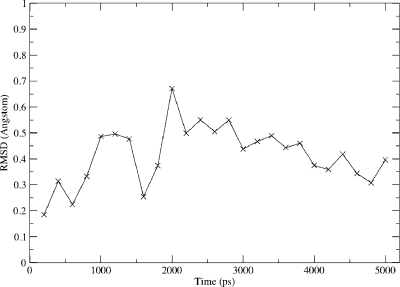
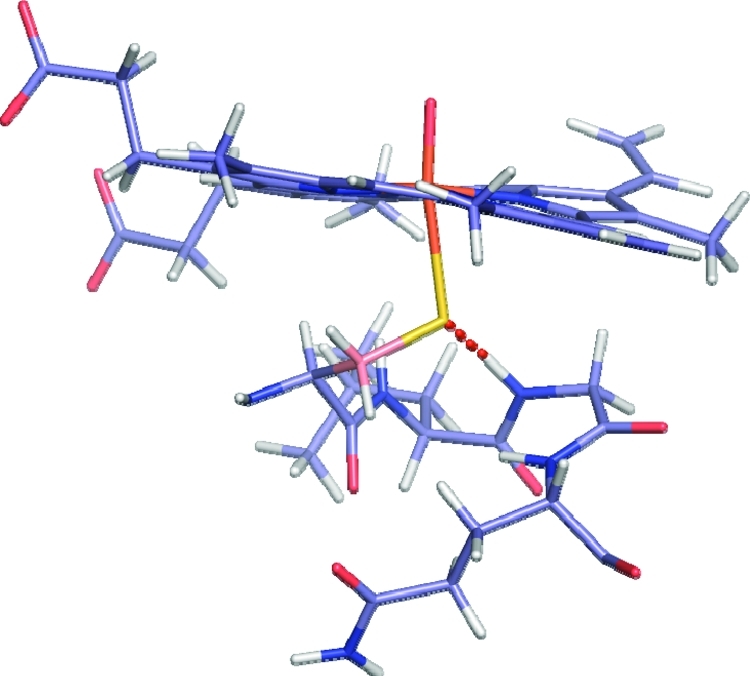
The cytochrome P450 (CYP) enzymes are important in many areas, including pharmaceutical development. Subtle changes in the electronic structure of the active species, Compound I, have been postulated previously to account partly for the experimentally observed differences in reactivity between isoforms. Current predictive models of CYP metabolism typically assume an identical Compound I in all isoforms. Here we present a method to calculate the electronic structure and to estimate the Fe-O bond enthalpy of Compound I, and apply it to several human and bacterial CYP isoforms. Conformational flexibility is accounted for by sampling large numbers of structures from molecular dynamics simulations, which are subsequently optimized with density functional theory (B3LYP) based quantum mechanics/molecular mechanics. The observed differences in Compound I between human isoforms are small: They are generally smaller than the spread of values obtained for the same isoform starting from different initial structures. Hence, it is unlikely that the variation in activity between human isoforms is due to differences in the electronic structure of Compound I. A larger difference in electronic structure is observed between the human isoforms and P450(cam) and may be explained by the slightly different hydrogen-bonding environment surrounding the cysteinyl sulfur. The presence of substrate in the active site of all isoforms studied appears to cause a slight decrease in the Fe-O bond enthalpy, apparently due to displacement of water out of the active site, suggesting that Compound I is less stable in the presence of substrate.
细胞色素 P450(CYP)酶在许多领域都很重要,包括药物开发。此前曾假设活性物质复合物 I 的电子结构发生微妙变化,部分解释了同工型之间反应性的实验观察差异。当前预测 CYP 代谢的模型通常假设所有同工型中都存在相同的复合物 I。在这里,我们提出了一种计算电子结构并估计复合物 I 的 Fe-O 键焓的方法,并将其应用于几种人类和细菌 CYP 同工型。构象灵活性通过从分子动力学模拟中采样大量结构来考虑,然后使用基于密度泛函理论(B3LYP)的量子力学/分子力学对其进行优化。在人类同工型之间观察到的复合物 I 差异较小:它们通常小于从不同初始结构获得的同一同工型的数值变化范围。因此,同工型之间活性的差异不太可能是由于复合物 I 的电子结构不同所致。在人类同工型和 P450(cam)之间观察到更大的电子结构差异,这可能是由于围绕半胱氨酸硫的氢键环境略有不同所致。在研究的所有同工型的活性位点中存在底物似乎会导致 Fe-O 键焓略有下降,这显然是由于活性位点中的水被排出,表明在底物存在下复合物 I 不太稳定。