Genopolis Consortium, University of Milano-Bicocca, Milan, 20126, Italy.
BMC Immunol. 2011 Aug 29;12:50. doi: 10.1186/1471-2172-12-50.
The selection of relevant genes for sample classification is a common task in many gene expression studies. Although a number of tools have been developed to identify optimal gene expression signatures, they often generate gene lists that are too long to be exploited clinically. Consequently, researchers in the field try to identify the smallest set of genes that provide good sample classification. We investigated the genome-wide expression of the inflammatory phenotype in dendritic cells. Dendritic cells are a complex group of cells that play a critical role in vertebrate immunity. Therefore, the prediction of the inflammatory phenotype in these cells may help with the selection of immune-modulating compounds.
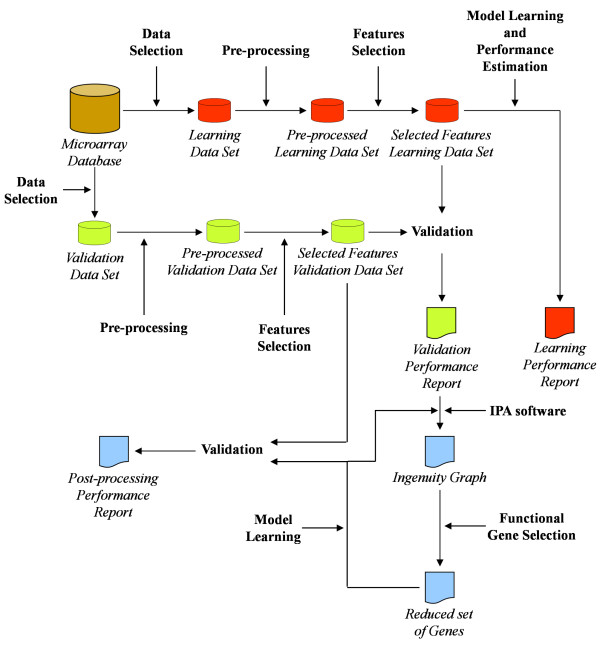
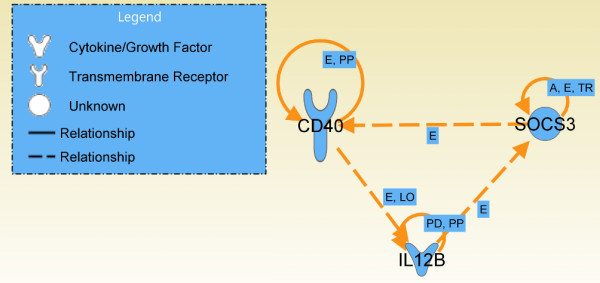
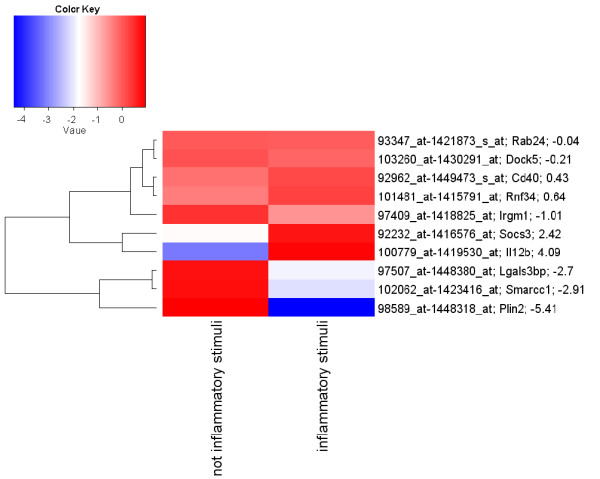
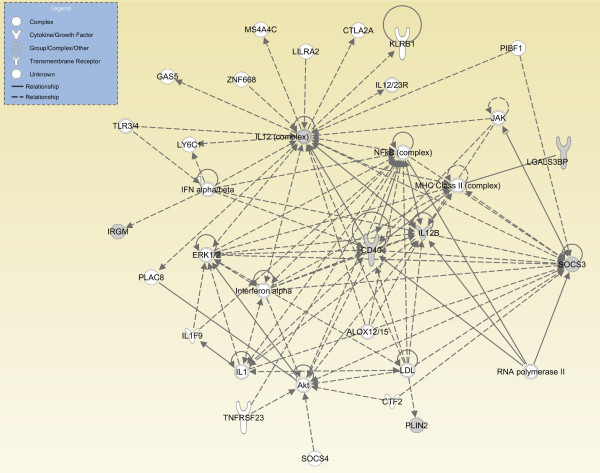
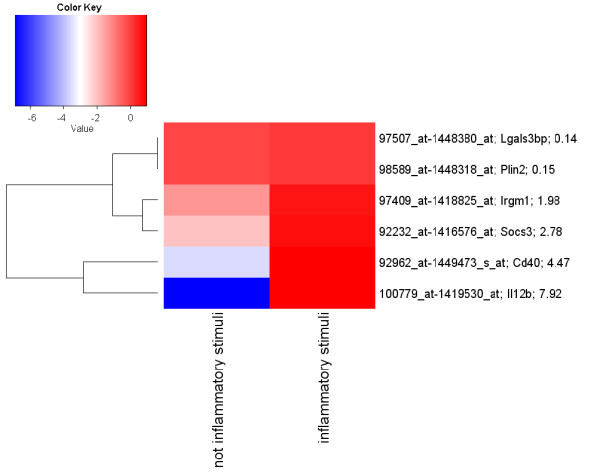
A data mining protocol was applied to microarray data for murine cell lines treated with various inflammatory stimuli. The learning and validation data sets consisted of 155 and 49 samples, respectively. The data mining protocol reduced the number of probe sets from 5,802 to 10, then from 10 to 6 and finally from 6 to 3. The performances of a set of supervised classification models were compared. The best accuracy, when using the six following genes --Il12b, Cd40, Socs3, Irgm1, Plin2 and Lgals3bp-- was obtained by Tree Augmented Naïve Bayes and Nearest Neighbour (91.8%). Using the smallest set of three genes --Il12b, Cd40 and Socs3-- the performance remained satisfactory and the best accuracy was with Support Vector Machine (95.9%). These data mining models, using data for the genes Il12b, Cd40 and Socs3, were validated with a human data set consisting of 27 samples. Support Vector Machines (71.4%) and Nearest Neighbour (92.6%) gave the worst performances, but the remaining models correctly classified all the 27 samples.
The genes selected by the data mining protocol proposed were shown to be informative for discriminating between inflammatory and steady-state phenotypes in dendritic cells. The robustness of the data mining protocol was confirmed by the accuracy for a human data set, when using only the following three genes: Il12b, Cd40 and Socs3. In summary, we analysed the longitudinal pattern of expression in dendritic cells stimulated with activating agents with the aim of identifying signatures that would predict or explain the dentritic cell response to an inflammatory agent.
选择与样本分类相关的基因是许多基因表达研究中的常见任务。虽然已经开发了许多工具来识别最佳的基因表达特征,但它们通常会生成过长的基因列表,难以在临床上应用。因此,该领域的研究人员试图确定提供良好样本分类的最小基因集。我们研究了树突状细胞炎症表型的全基因组表达。树突状细胞是一组复杂的细胞,在脊椎动物免疫中起着关键作用。因此,预测这些细胞的炎症表型可能有助于选择免疫调节化合物。
应用于经各种炎症刺激处理的鼠系细胞的微阵列数据的数据挖掘方案。学习和验证数据集分别包含 155 和 49 个样本。数据挖掘方案将探针集的数量从 5802 减少到 10,然后从 10 减少到 6,最后从 6 减少到 3。比较了一组监督分类模型的性能。当使用以下六个基因(Il12b、Cd40、Socs3、Irgm1、Plin2 和 Lgals3bp)时,Tree Augmented Naive Bayes 和 Nearest Neighbour 的准确性最高(91.8%)。使用三个基因(Il12b、Cd40 和 Socs3)的最小集合,性能仍然令人满意,支持向量机的准确性最高(95.9%)。使用基因 Il12b、Cd40 和 Socs3 的这些数据挖掘模型,使用包含 27 个样本的人类数据集进行了验证。支持向量机(71.4%)和最近邻(92.6%)的性能最差,但其余模型正确分类了所有 27 个样本。
所提出的数据挖掘方案选择的基因被证明可用于区分树突状细胞的炎症和稳态表型。当仅使用以下三个基因(Il12b、Cd40 和 Socs3)时,对人类数据集的准确性证实了数据挖掘方案的稳健性。总之,我们分析了用激活剂刺激的树突状细胞的纵向表达模式,目的是确定可预测或解释树突状细胞对炎症剂反应的特征。