Department of Pharmacology, University of Tennessee Health Science Center Memphis, TN, USA.
Front Neurosci. 2011 Aug 12;5:98. doi: 10.3389/fnins.2011.00098. eCollection 2011.
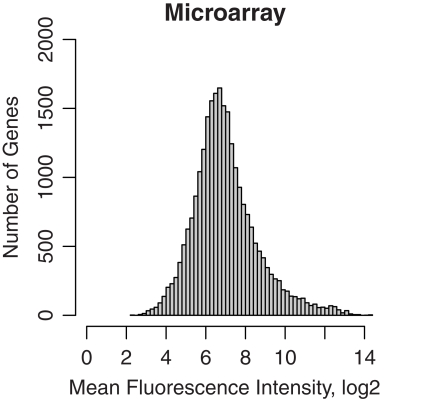
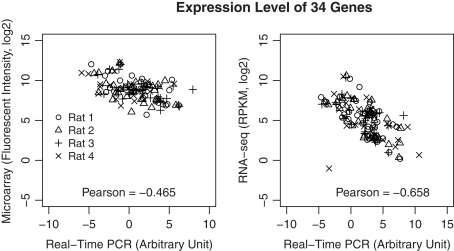
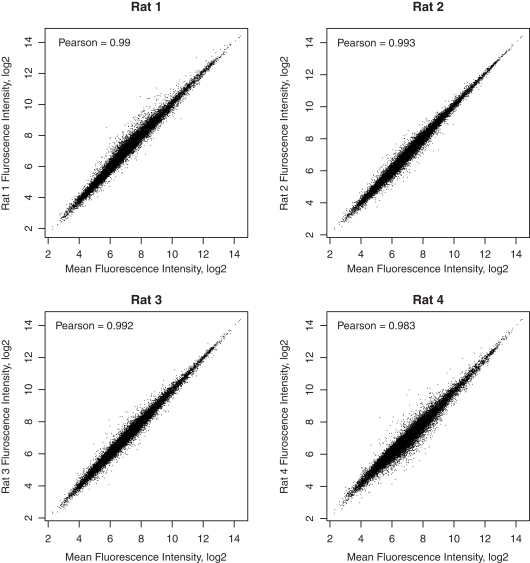
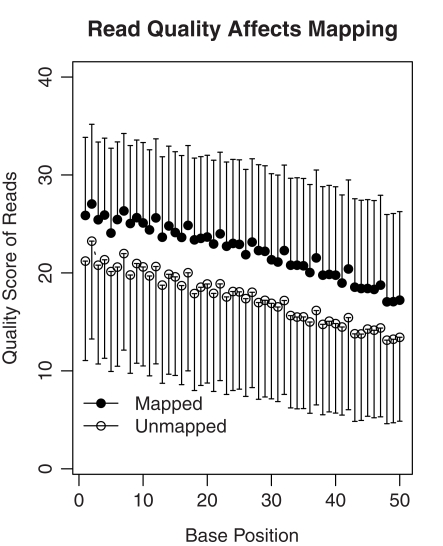
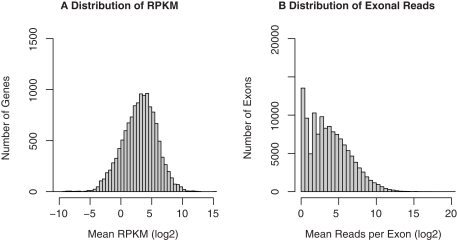
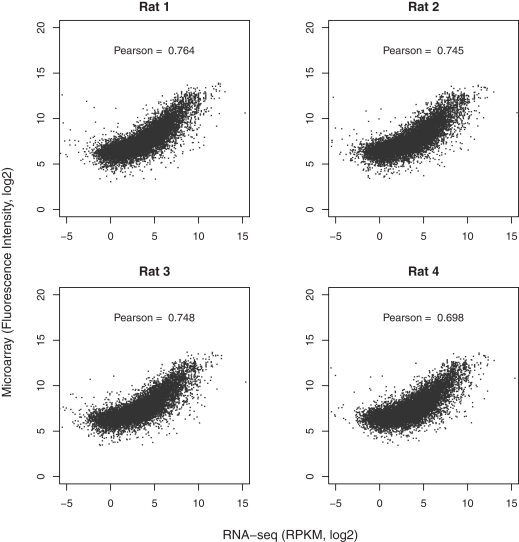
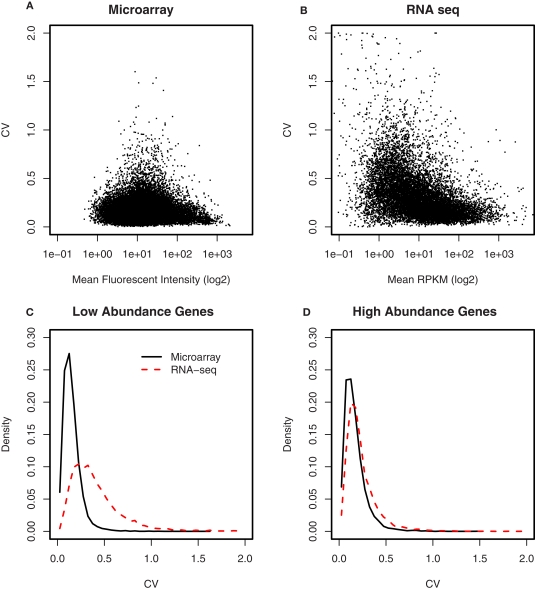
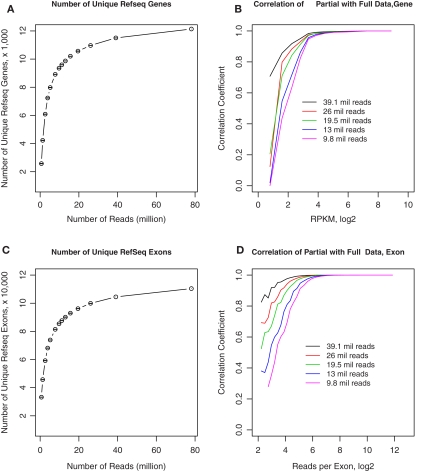
The cellular heterogeneity of brain poses a particularly thorny issue in genome-wide gene expression studies. Because laser capture microdissection (LCM) enables the precise extraction of a small area of tissue, we combined LCM with neuronal track tracing to collect nucleus accumbens shell neurons that project to ventral pallidum, which are of particular interest in the study of reward and addiction. Four independent biological samples of accumbens projection neurons were obtained. Approximately 500 pg of total RNA from each sample was then amplified linearly and subjected to Affymetrix microarray and Applied Biosystems sequencing by oligonucleotide ligation and detection (SOLiD) transcriptome sequencing (RNA-seq). A total of 375 million 50-bp reads were obtained from RNA-seq. Approximately 57% of these reads were mapped to the rat reference genome (Baylor 3.4/rn4). Approximately 11,000 unique RefSeq genes and 100,000 unique exons were identified from each sample. Of the unmapped reads, the quality scores were 4.74 ± 0.42 lower than the mapped reads. When RNA-seq and microarray data from the same samples were compared, Pearson correlations were between 0.764 and 0.798. The variances in data obtained for the four samples by microarray and RNA-seq were similar for medium to high abundance genes, but less among low abundance genes detected by microarray. Analysis of 34 genes by real-time polymerase chain reaction showed higher correlation with RNA-seq (0.66) than with microarray (0.46). Further analysis showed 20-30 million 50-bp reads are sufficient to provide estimates of gene expression levels comparable to those produced by microarray. In summary, this study showed that picogram quantities of total RNA obtained by LCM of ∼700 individual neurons is sufficient to take advantage of the benefits provided by the transcriptome sequencing technology, such as low background noise, high dynamic range, and high precision.
大脑的细胞异质性在全基因组基因表达研究中构成了一个特别棘手的问题。由于激光捕获显微切割(LCM)能够精确地提取一小部分组织,因此我们将 LCM 与神经元追踪相结合,以收集投射到伏隔核苍白球的壳状神经元,这些神经元在研究奖励和成瘾方面特别有趣。获得了四个独立的 Accumbens 投射神经元的生物样本。然后,从每个样本中线性扩增约 500pg 的总 RNA,并进行 Affymetrix 微阵列和 Applied Biosystems 测序通过寡核苷酸连接和检测(SOLiD)转录组测序(RNA-seq)。从 RNA-seq 获得了总共 3.75 亿个 50bp 读取。这些读取中约有 57%映射到大鼠参考基因组(Baylor 3.4/rn4)。从每个样本中鉴定出约 11000 个独特的 RefSeq 基因和 100000 个独特的外显子。未映射的读取的质量分数比映射的读取低 4.74±0.42。当比较来自相同样本的 RNA-seq 和微阵列数据时,Pearson 相关系数在 0.764 到 0.798 之间。对于微阵列和 RNA-seq 获得的四个样本的数据,中高丰度基因的方差相似,但微阵列检测到的低丰度基因的方差较小。通过实时聚合酶链反应分析 34 个基因,与 RNA-seq 的相关性更高(0.66),与微阵列的相关性较低(0.46)。进一步分析表明,2000 万到 3000 万个 50bp 读取足以提供与微阵列相当的基因表达水平估计。总之,这项研究表明,通过 LCM 从约 700 个单个神经元中获得的皮克数量的总 RNA 足以利用转录组测序技术提供的好处,例如背景噪声低、动态范围宽和精度高。