Department of Microbiology and Immunology, University of British Columbia, Vancouver, British Columbia, Canada.
PLoS One. 2011;6(8):e23789. doi: 10.1371/journal.pone.0023789. Epub 2011 Aug 31.
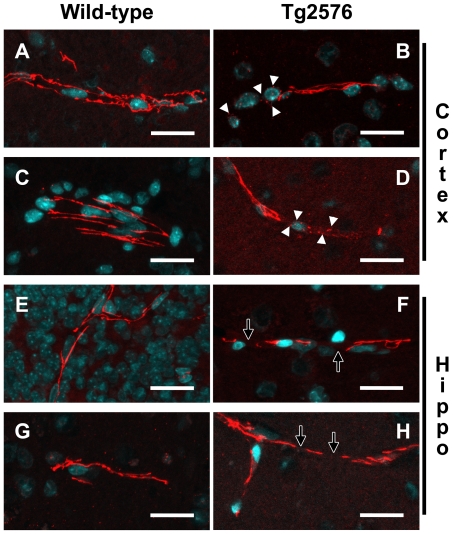
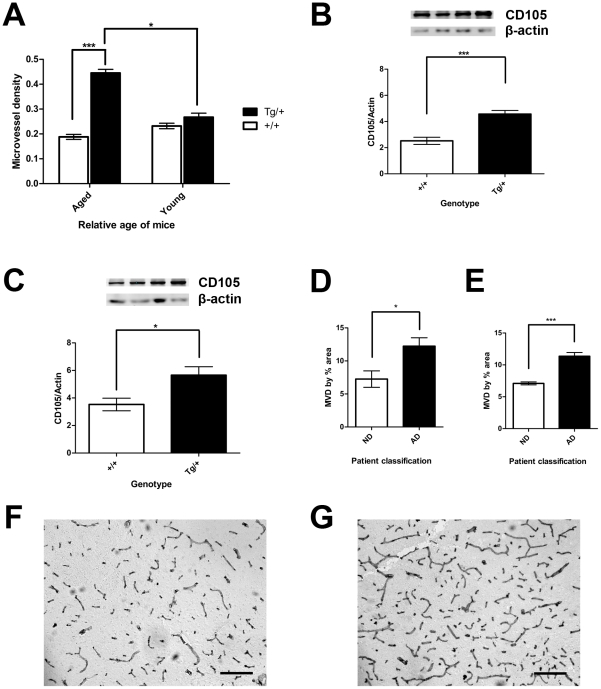
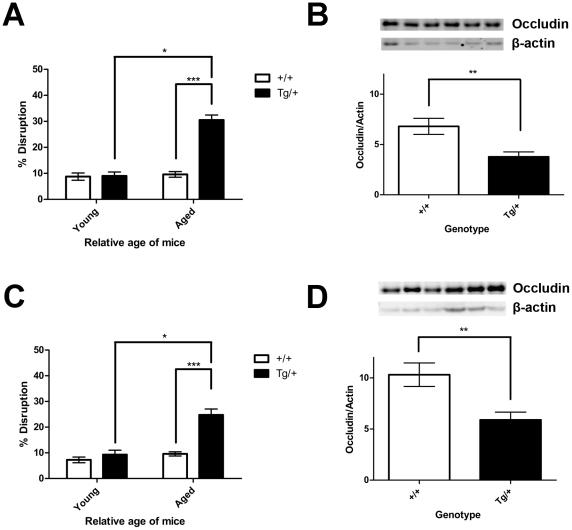
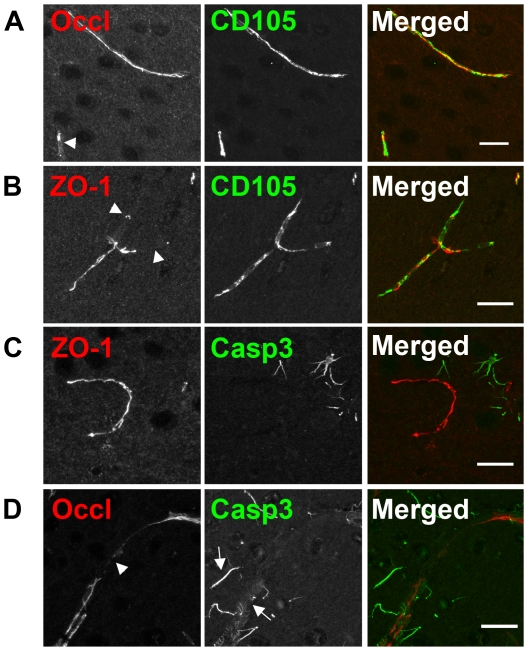
Evidence of reduced blood-brain barrier (BBB) integrity preceding other Alzheimer's disease (AD) pathology provides a strong link between cerebrovascular angiopathy and AD. However, the "Vascular hypothesis", holds that BBB leakiness in AD is likely due to hypoxia and neuroinflammation leading to vascular deterioration and apoptosis. We propose an alternative hypothesis: amyloidogenesis promotes extensive neoangiogenesis leading to increased vascular permeability and subsequent hypervascularization in AD. Cerebrovascular integrity was characterized in Tg2576 AD model mice that overexpress the human amyloid precursor protein (APP) containing the double missense mutations, APPsw, found in a Swedish family, that causes early-onset AD. The expression of tight junction (TJ) proteins, occludin and ZO-1, were examined in conjunction with markers of apoptosis and angiogenesis. In aged Tg2576 AD mice, a significant increase in the incidence of disrupted TJs, compared to age matched wild-type littermates and young mice of both genotypes, was directly linked to an increased microvascular density but not apoptosis, which strongly supports amyloidogenic triggered hypervascularity as the basis for BBB disruption. Hypervascularity in human patients was corroborated in a comparison of postmortem brain tissues from AD and controls. Our results demonstrate that amylodogenesis mediates BBB disruption and leakiness through promoting neoangiogenesis and hypervascularity, resulting in the redistribution of TJs that maintain the barrier and thus, provides a new paradigm for integrating vascular remodeling with the pathophysiology observed in AD. Thus the extensive angiogenesis identified in AD brain, exhibits parallels to the neovascularity evident in the pathophysiology of other diseases such as age-related macular degeneration.
有证据表明,血脑屏障(BBB)完整性的降低先于其他阿尔茨海默病(AD)病理学,这为脑血管病和 AD 之间提供了一个强有力的联系。然而,“血管假说”认为,AD 中 BBB 的渗漏很可能是由于缺氧和神经炎症导致血管恶化和细胞凋亡。我们提出了一个替代假说:淀粉样蛋白的形成促进了广泛的新生血管形成,导致 AD 中血管通透性增加和随后的过度血管化。我们在过表达人类淀粉样前体蛋白(APP)的 Tg2576 AD 模型小鼠中研究了脑血管完整性,该蛋白含有在一个瑞典家族中发现的导致早发性 AD 的双错义突变 APPsw。紧密连接(TJ)蛋白、occludin 和 ZO-1 的表达与细胞凋亡和血管生成的标志物一起进行了检查。在老年 Tg2576 AD 小鼠中,与年龄匹配的野生型同窝仔鼠和两种基因型的年轻小鼠相比,TJ 破坏的发生率显著增加,这与微血管密度的增加直接相关,但与细胞凋亡无关,这强烈支持淀粉样蛋白触发的过度血管生成是 BBB 破坏的基础。在 AD 和对照组死后脑组织的比较中,证实了人类患者的过度血管生成。我们的研究结果表明,淀粉样蛋白的形成通过促进新生血管生成和过度血管化介导了 BBB 的破坏和渗漏,导致维持屏障的 TJ 重新分布,从而为将血管重塑与 AD 中观察到的病理生理学相结合提供了一个新的范例。因此,在 AD 大脑中发现的广泛血管生成与其他疾病(如年龄相关性黄斑变性)的病理生理学中明显的新生血管生成具有相似之处。