Institute of Microbial Technology, Chandigarh, India.
PLoS One. 2011;6(9):e24039. doi: 10.1371/journal.pone.0024039. Epub 2011 Sep 13.
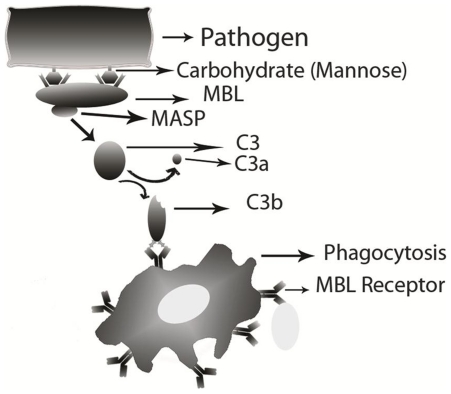
Mannose binding proteins (MBPs) play a vital role in several biological functions such as defense mechanisms. These proteins bind to mannose on the surface of a wide range of pathogens and help in eliminating these pathogens from our body. Thus, it is important to identify mannose interacting residues (MIRs) in order to understand mechanism of recognition of pathogens by MBPs.
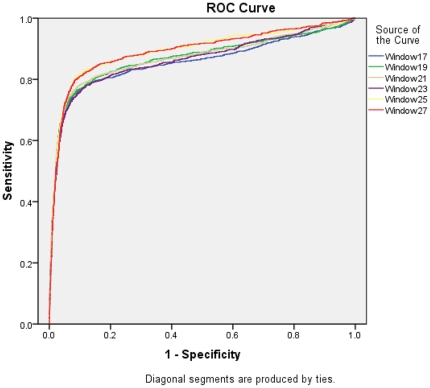
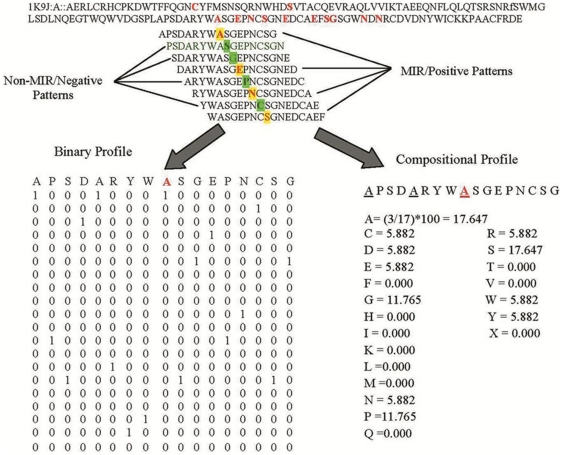
This paper describes modules developed for predicting MIRs in a protein. Support vector machine (SVM) based models have been developed on 120 mannose binding protein chains, where no two chains have more than 25% sequence similarity. SVM models were developed on two types of datasets: 1) main dataset consists of 1029 mannose interacting and 1029 non-interacting residues, 2) realistic dataset consists of 1029 mannose interacting and 10320 non-interacting residues. In this study, firstly, we developed standard modules using binary and PSSM profile of patterns and got maximum MCC around 0.32. Secondly, we developed SVM modules using composition profile of patterns and achieved maximum MCC around 0.74 with accuracy 86.64% on main dataset. Thirdly, we developed a model on a realistic dataset and achieved maximum MCC of 0.62 with accuracy 93.08%. Based on this study, a standalone program and web server have been developed for predicting mannose interacting residues in proteins (http://www.imtech.res.in/raghava/premier/).
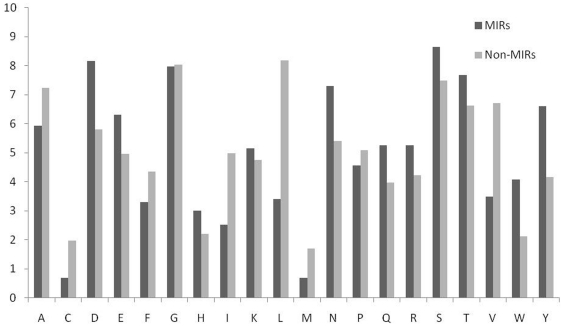
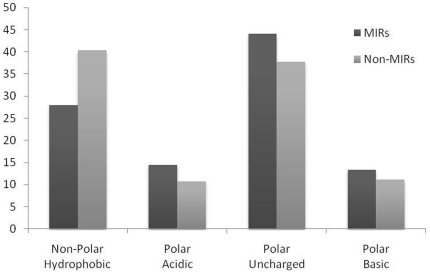
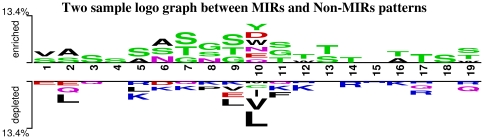
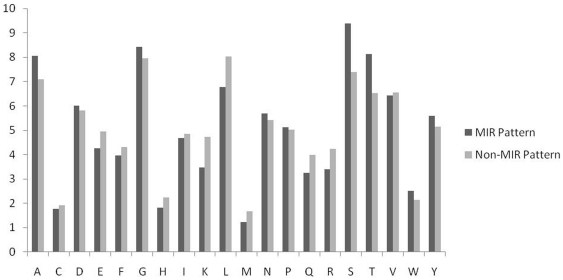
Compositional analysis of mannose interacting and non-interacting residues shows that certain types of residues are preferred in mannose interaction. It was also observed that residues around mannose interacting residues have a preference for certain types of residues. Composition of patterns/peptide/segment has been used for predicting MIRs and achieved reasonable high accuracy. It is possible that this novel strategy may be effective to predict other types of interacting residues. This study will be useful in annotating the function of protein as well as in understanding the role of mannose in the immune system.
甘露糖结合蛋白(MBP)在多种生物学功能中发挥着重要作用,如防御机制。这些蛋白质与多种病原体表面的甘露糖结合,有助于清除体内的这些病原体。因此,识别甘露糖相互作用残基(MIR)对于了解 MBP 识别病原体的机制非常重要。
本文描述了用于预测蛋白质中 MIR 的模块。基于支持向量机(SVM)的模型已经在 120 个甘露糖结合蛋白链上开发,其中没有两个链具有超过 25%的序列相似性。SVM 模型是在两种类型的数据集上开发的:1)主要数据集由 1029 个甘露糖相互作用残基和 1029 个非相互作用残基组成,2)实际数据集由 1029 个甘露糖相互作用残基和 10320 个非相互作用残基组成。在这项研究中,首先,我们使用二元和 PSSM 模式的图案以及最大 MCC 约为 0.32 来开发标准模块。其次,我们使用模式的组成图案来开发 SVM 模块,在主要数据集上实现了最大 MCC 约 0.74,准确率为 86.64%。第三,我们在实际数据集上开发了一个模型,最大 MCC 为 0.62,准确率为 93.08%。基于这项研究,我们开发了一个用于预测蛋白质中甘露糖相互作用残基的独立程序和网络服务器(http://www.imtech.res.in/raghava/premier/)。
甘露糖相互作用和非相互作用残基的组成分析表明,某些类型的残基在甘露糖相互作用中是优先的。还观察到,甘露糖相互作用残基周围的残基对某些类型的残基有偏好。模式/肽/段的组成已被用于预测 MIR,并取得了相当高的准确性。这种新策略可能对预测其他类型的相互作用残基有效。这项研究将有助于注释蛋白质的功能以及理解甘露糖在免疫系统中的作用。