Institute of Microbial Technology, Chandigarh, India.
BMC Bioinformatics. 2009 Dec 19;10:434. doi: 10.1186/1471-2105-10-434.
One of the major challenges in post-genomic era is to provide functional annotations for large number of proteins arising from genome sequencing projects. The function of many proteins depends on their interaction with small molecules or ligands. ATP is one such important ligand that plays critical role as a coenzyme in the functionality of many proteins. There is a need to develop method for identifying ATP interacting residues in a ATP binding proteins (ABPs), in order to understand mechanism of protein-ligands interaction.
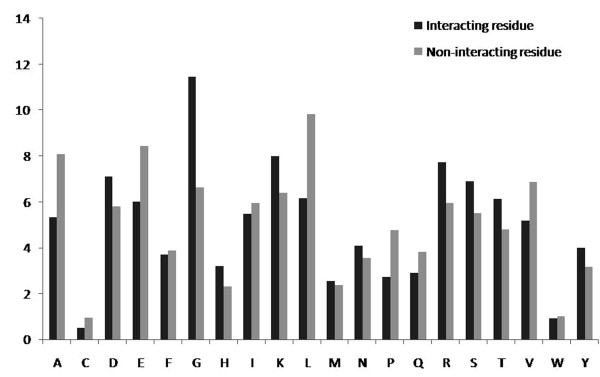
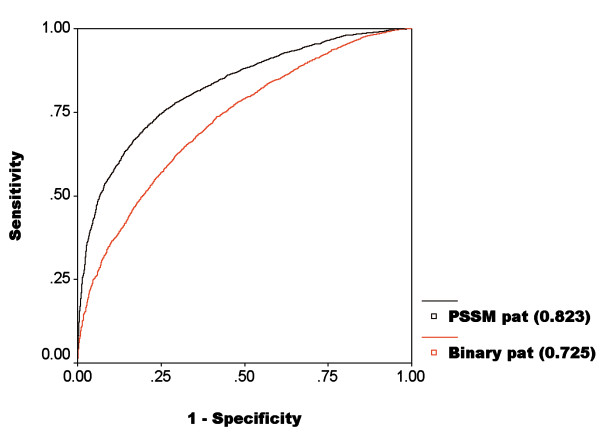
We have compared the amino acid composition of ATP interacting and non-interacting regions of proteins and observed that certain residues are preferred for interaction with ATP. This study describes few models that have been developed for identifying ATP interacting residues in a protein. All these models were trained and tested on 168 non-redundant ABPs chains. First we have developed a Support Vector Machine (SVM) based model using primary sequence of proteins and obtained maximum MCC 0.33 with accuracy of 66.25%. Secondly, another SVM based model was developed using position specific scoring matrix (PSSM) generated by PSI-BLAST. The performance of this model was improved significantly (MCC 0.5) from the previous one, where only the primary sequence of the proteins were used.
This study demonstrates that it is possible to predict 'ATP interacting residues' in a protein with moderate accuracy using its sequence. The evolutionary information is important for the identification of 'ATP interacting residues', as it provides more information compared to the primary sequence. This method will be useful for researchers studying ATP-binding proteins. Based on this study, a web server has been developed for predicting 'ATP interacting residues' in a protein http://www.imtech.res.in/raghava/atpint/.
在后基因组时代,主要挑战之一是为基因组测序项目产生的大量蛋白质提供功能注释。许多蛋白质的功能取决于它们与小分子或配体的相互作用。ATP 就是这样一种重要的配体,它作为许多蛋白质功能的辅酶起着至关重要的作用。因此,需要开发一种识别 ATP 结合蛋白 (ABP) 中与 ATP 相互作用的残基的方法,以便了解蛋白质-配体相互作用的机制。
我们比较了蛋白质中与 ATP 相互作用和非相互作用区域的氨基酸组成,观察到某些残基更倾向于与 ATP 相互作用。本研究描述了几种已开发用于识别蛋白质中与 ATP 相互作用的残基的模型。所有这些模型都是在 168 个非冗余 ABP 链上进行训练和测试的。首先,我们使用蛋白质的一级序列开发了基于支持向量机 (SVM) 的模型,得到了最大 MCC 为 0.33,准确率为 66.25%。其次,我们使用 PSI-BLAST 生成的位置特异性评分矩阵 (PSSM) 开发了另一个基于 SVM 的模型。与仅使用蛋白质一级序列的前一个模型相比,该模型的性能有了显著提高(MCC 为 0.5)。
本研究表明,使用其序列以中等精度预测蛋白质中的“ATP 相互作用残基”是可能的。进化信息对于鉴定“ATP 相互作用残基”很重要,因为它提供的信息比一级序列更多。该方法将对研究 ATP 结合蛋白的研究人员有用。基于这项研究,我们开发了一个用于预测蛋白质中“ATP 相互作用残基”的网络服务器:http://www.imtech.res.in/raghava/atpint/。