Institute of Microbial Technology, Sector 39A, Chandigarh, 160036, India.
BMC Bioinformatics. 2010 Mar 30;11:160. doi: 10.1186/1471-2105-11-160.
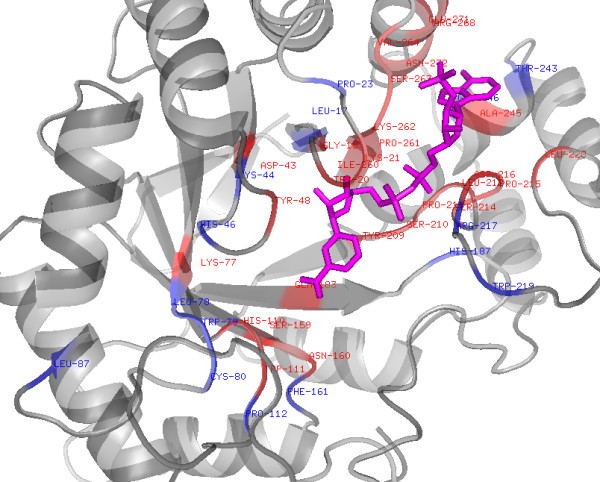
Small molecular cofactors or ligands play a crucial role in the proper functioning of cells. Accurate annotation of their target proteins and binding sites is required for the complete understanding of reaction mechanisms. Nicotinamide adenine dinucleotide (NAD+ or NAD) is one of the most commonly used organic cofactors in living cells, which plays a critical role in cellular metabolism, storage and regulatory processes. In the past, several NAD binding proteins (NADBP) have been reported in the literature, which are responsible for a wide-range of activities in the cell. Attempts have been made to derive a rule for the binding of NAD+ to its target proteins. However, so far an efficient model could not be derived due to the time consuming process of structure determination, and limitations of similarity based approaches. Thus a sequence and non-similarity based method is needed to characterize the NAD binding sites to help in the annotation. In this study attempts have been made to predict NAD binding proteins and their interacting residues (NIRs) from amino acid sequence using bioinformatics tools.
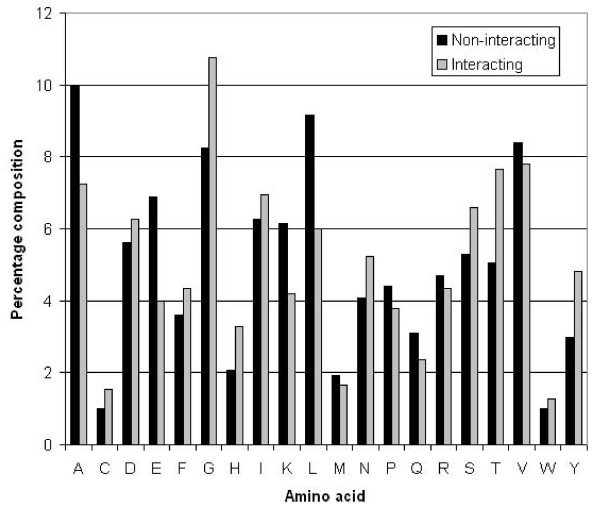
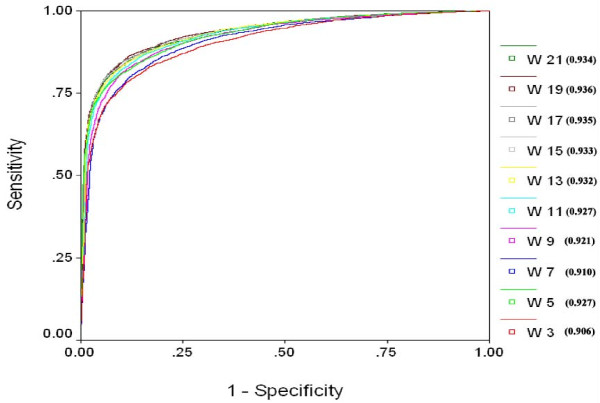
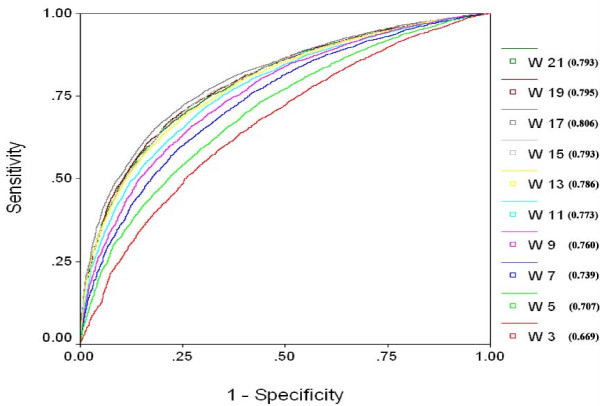
We extracted 1556 proteins chains from 555 NAD binding proteins whose structure is available in Protein Data Bank. Then we removed all redundant protein chains and finally obtained 195 non-redundant NAD binding protein chains, where no two chains have more than 40% sequence identity. In this study all models were developed and evaluated using five-fold cross validation technique on the above dataset of 195 NAD binding proteins. While certain type of residues are preferred (e.g. Gly, Tyr, Thr, His) in NAD interaction, residues like Ala, Glu, Leu, Lys are not preferred. A support vector machine (SVM) based method has been developed using various window lengths of amino acid sequence for predicting NAD interacting residues and obtained maximum Matthew's correlation coefficient (MCC) 0.47 with accuracy 74.13% at window length 17. We also developed a SVM based method using evolutionary information in the form of position specific scoring matrix (PSSM) and obtained maximum MCC 0.75 with accuracy 87.25%.
For the first time a sequence-based method has been developed for the prediction of NAD binding proteins and their interacting residues, in the absence of any prior structural information. The present model will aid in the understanding of NAD+ dependent mechanisms of action in the cell. To provide service to the scientific community, we have developed a user-friendly web server, which is available from URL http://www.imtech.res.in/raghava/nadbinder/.
小分子辅因子或配体在细胞的正常功能中起着至关重要的作用。为了完全了解反应机制,需要准确注释其靶蛋白和结合位点。烟酰胺腺嘌呤二核苷酸(NAD+或 NAD)是活细胞中最常用的有机辅因子之一,它在细胞代谢、储存和调节过程中起着关键作用。过去,文献中已经报道了几种 NAD 结合蛋白(NADBP),它们负责细胞中的广泛活动。已经尝试得出 NAD+与其靶蛋白结合的规则。然而,到目前为止,由于结构确定过程耗时且基于相似性的方法存在局限性,因此无法得出有效的模型。因此,需要一种基于序列和非相似性的方法来描述 NAD 结合位点,以帮助注释。在这项研究中,我们尝试使用生物信息学工具从氨基酸序列预测 NAD 结合蛋白及其相互作用残基(NIRs)。
我们从蛋白质数据库中提取了结构可用的 555 种 NAD 结合蛋白的 1556 个蛋白质链。然后,我们去除了所有冗余的蛋白质链,最后得到了 195 个非冗余的 NAD 结合蛋白链,其中没有两条链的序列同一性超过 40%。在这项研究中,所有模型都是使用上述 195 种 NAD 结合蛋白数据集的五重交叉验证技术开发和评估的。虽然在 NAD 相互作用中某些类型的残基(例如 Gly、Tyr、Thr、His)是首选的,但残基(如 Ala、Glu、Leu、Lys)则不是首选的。我们使用各种氨基酸序列窗口长度开发了基于支持向量机(SVM)的方法来预测 NAD 相互作用残基,并在窗口长度为 17 时获得了最大马修相关系数(MCC)0.47 和准确率 74.13%。我们还使用位置特定评分矩阵(PSSM)的形式开发了基于 SVM 的方法,并获得了最大 MCC 0.75 和准确率 87.25%。
这是首次在没有任何先前结构信息的情况下,基于序列开发了预测 NAD 结合蛋白及其相互作用残基的方法。该模型将有助于理解 NAD+在细胞中的作用机制。为了为科学界提供服务,我们开发了一个用户友好的网络服务器,可从 URL http://www.imtech.res.in/raghava/nadbinder/ 访问。