Joint BSC-IRB research programme in Computational Biology, Barcelona Supercomputing Center, Barcelona 08034, Spain.
BMC Bioinformatics. 2011 Sep 26;12:378. doi: 10.1186/1471-2105-12-378.
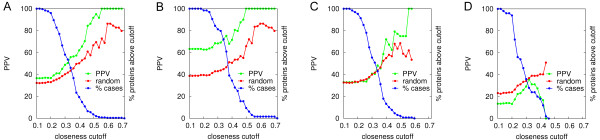
Protein-protein interactions are involved in most cellular processes, and their detailed physico-chemical and structural characterization is needed in order to understand their function at the molecular level. In-silico docking tools can complement experimental techniques, providing three-dimensional structural models of such interactions at atomic resolution. In several recent studies, protein structures have been modeled as networks (or graphs), where the nodes represent residues and the connecting edges their interactions. From such networks, it is possible to calculate different topology-based values for each of the nodes, and to identify protein regions with high centrality scores, which are known to positively correlate with key functional residues, hot spots, and protein-protein interfaces.
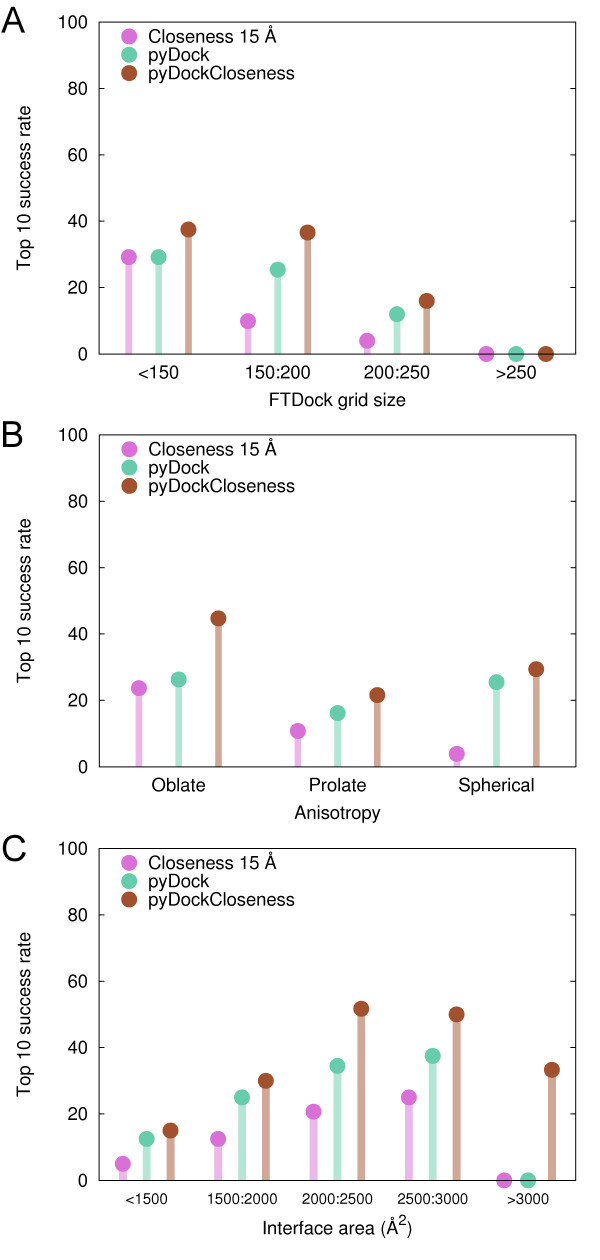
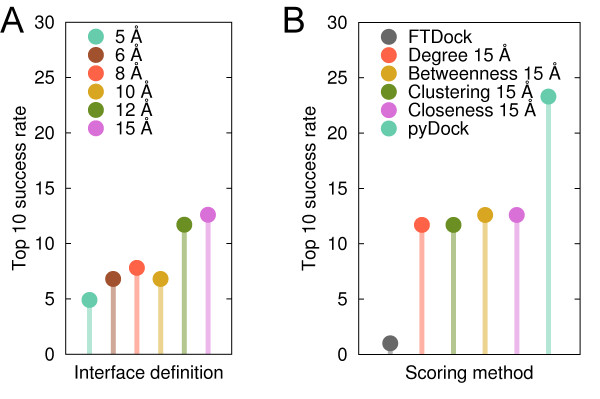
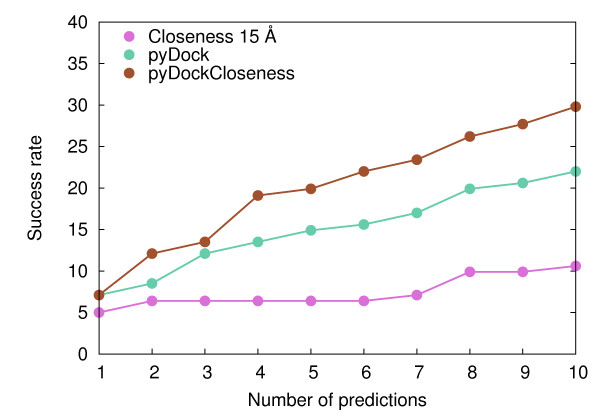
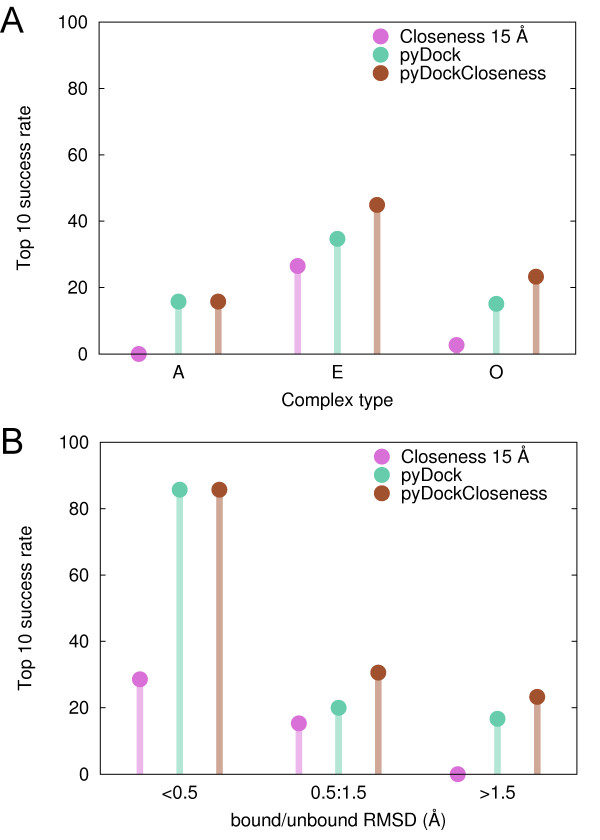
Here we show that this correlation can be efficiently used for the scoring of rigid-body docking poses. When integrated into the pyDock energy-based docking method, the new combined scoring function significantly improved the results of the individual components as shown on a standard docking benchmark. This improvement was particularly remarkable for specific protein complexes, depending on the shape, size, type, or flexibility of the proteins involved.
The network-based representation of protein structures can be used to identify protein-protein binding regions and to efficiently score docking poses, complementing energy-based approaches.
蛋白质-蛋白质相互作用涉及大多数细胞过程,为了在分子水平上理解它们的功能,需要对其详细的物理化学和结构特征进行描述。在计算机上进行对接工具可以补充实验技术,提供这些相互作用的原子分辨率的三维结构模型。在最近的几项研究中,蛋白质结构被建模为网络(或图),其中节点代表残基,连接的边代表它们的相互作用。从这样的网络中,可以为每个节点计算不同的基于拓扑的值,并确定具有高中心性得分的蛋白质区域,这些区域与关键功能残基、热点和蛋白质-蛋白质界面呈正相关。
在这里,我们表明这种相关性可以有效地用于刚体对接构象的评分。当集成到基于能量的 pyDock 对接方法中时,新的组合评分函数显著提高了标准对接基准测试中各个组件的结果。对于特定的蛋白质复合物,这种改进特别显著,具体取决于涉及的蛋白质的形状、大小、类型或灵活性。
蛋白质结构的基于网络的表示可以用于识别蛋白质-蛋白质结合区域,并有效地对对接构象进行评分,补充基于能量的方法。