Malaria Branch, Division of Parasitic Diseases and Malaria, Center for Global Health, Centers for Disease Control and Prevention, Atlanta, Georgia, United States of America.
PLoS One. 2011;6(9):e23486. doi: 10.1371/journal.pone.0023486. Epub 2011 Sep 16.
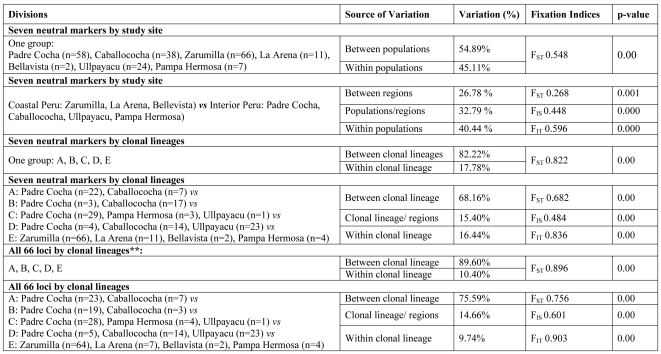
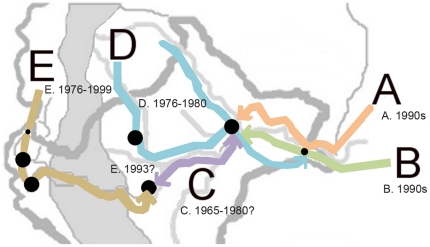
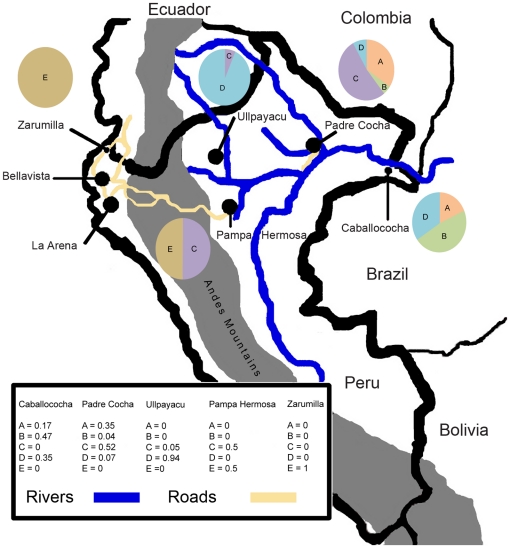
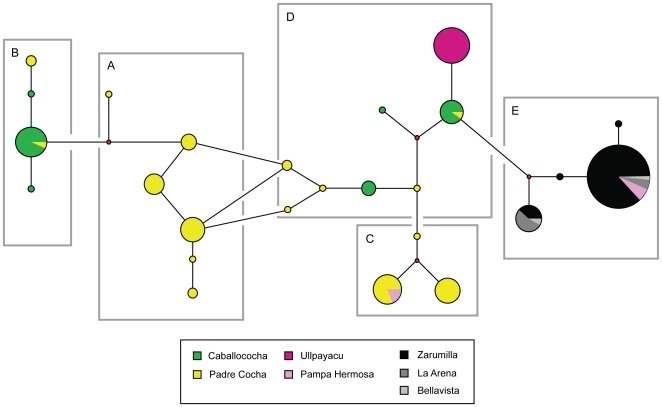
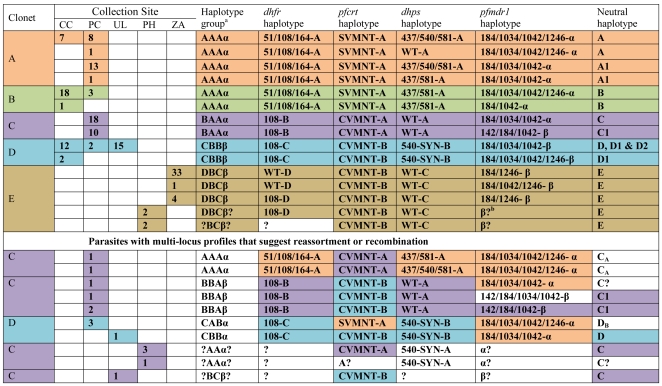
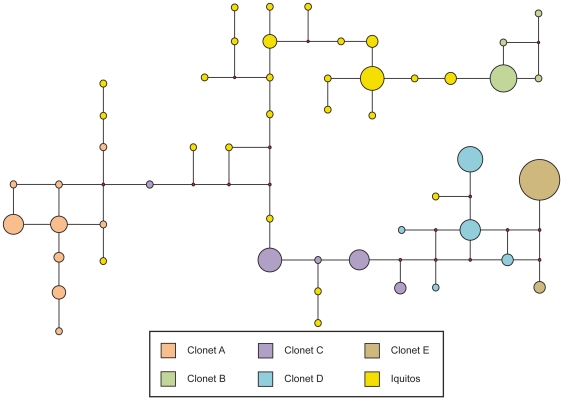
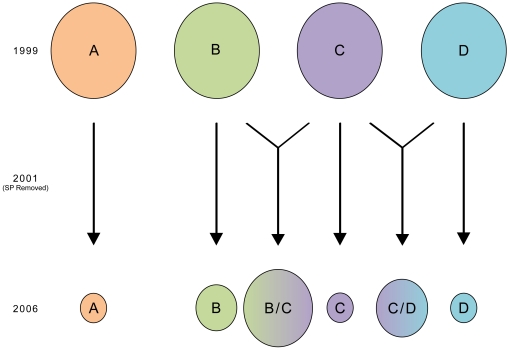
Malaria has reemerged in many regions where once it was nearly eliminated. Yet the source of these parasites, the process of repopulation, their population structure, and dynamics are ill defined. Peru was one of malaria eradication's successes, where Plasmodium falciparum was nearly eliminated for two decades. It reemerged in the 1990s. In the new era of malaria elimination, Peruvian P. falciparum is a model of malaria reinvasion. We investigated its population structure and drug resistance profiles. We hypothesized that only populations adapted to local ecological niches could expand and repopulate and originated as vestigial populations or recent introductions. We investigated the genetic structure (using microsatellites) and drug resistant genotypes of 220 parasites collected from patients immediately after peak epidemic expansion (1999-2000) from seven sites across the country. The majority of parasites could be grouped into five clonal lineages by networks and AMOVA. The distribution of clonal lineages and their drug sensitivity profiles suggested geographic structure. In 2001, artesunate combination therapy was introduced in Peru. We tested 62 parasites collected in 2006-2007 for changes in genetic structure. Clonal lineages had recombined under selection for the fittest parasites. Our findings illustrate that local adaptations in the post-eradication era have contributed to clonal lineage expansion. Within the shifting confluence of drug policy and malaria incidence, populations continue to evolve through genetic outcrossing influenced by antimalarial selection pressure. Understanding the population substructure of P. falciparum has implications for vaccine, drug, and epidemiologic studies, including monitoring malaria during and after the elimination phase.
疟疾在许多曾经几乎被消灭的地区重新出现。然而,这些寄生虫的来源、重新繁殖的过程、它们的种群结构和动态仍然不清楚。秘鲁是消灭疟疾的成功案例之一,在过去的二十年里,恶性疟原虫几乎被消灭。它在 20 世纪 90 年代重新出现。在消除疟疾的新时代,秘鲁的恶性疟原虫是疟疾重新入侵的一个模型。我们研究了它的种群结构和耐药性特征。我们假设,只有适应当地生态位的种群才能扩张和重新繁殖,并起源于残余种群或最近的引入。我们研究了从全国七个地点在流行高峰期(1999-2000 年)后立即收集的 220 个寄生虫的遗传结构(使用微卫星)和耐药基因型。大多数寄生虫可以通过网络和 AMOVA 将其分为五个克隆谱系。克隆谱系的分布及其耐药基因型表明存在地理结构。2001 年,在秘鲁引入了青蒿琥酯联合疗法。我们测试了 2006-2007 年收集的 62 个寄生虫,以了解遗传结构的变化。在适合的寄生虫选择下,克隆谱系发生了重组。我们的研究结果表明,在消灭后时代,局部适应有助于克隆谱系的扩张。在药物政策和疟疾发病率的不断变化的汇合中,通过抗疟选择压力影响的遗传杂交,种群继续进化。了解恶性疟原虫的种群亚结构对疫苗、药物和流行病学研究具有重要意义,包括在消灭阶段及其后监测疟疾。