Department of Computer Science, Virginia Polytechnic Institute and State University, Blacksburg, Virginia, United States of America.
PLoS Comput Biol. 2011 Sep;7(9):e1002164. doi: 10.1371/journal.pcbi.1002164. Epub 2011 Sep 22.
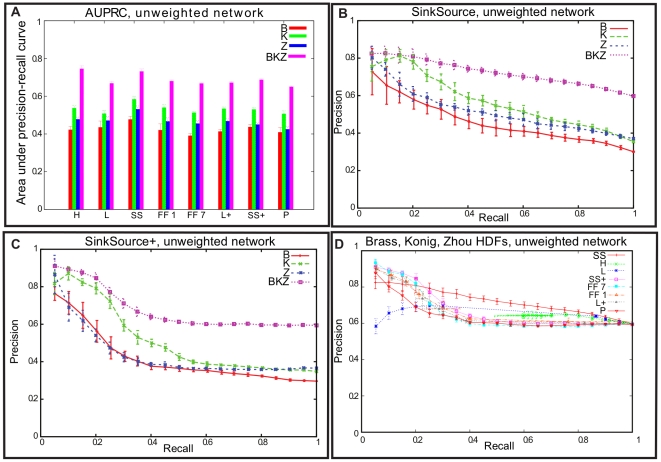
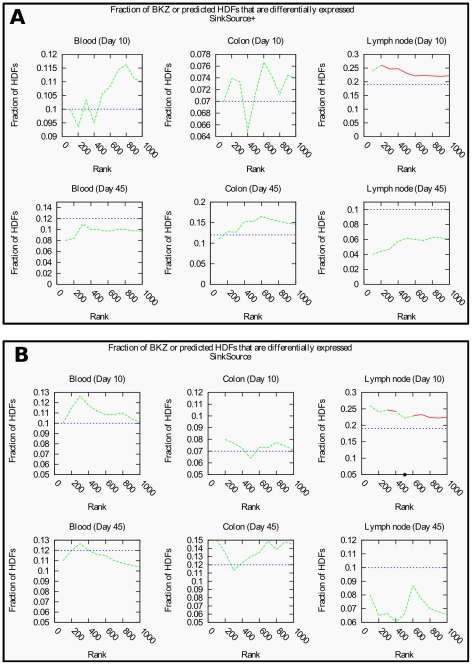
HIV Dependency Factors (HDFs) are a class of human proteins that are essential for HIV replication, but are not lethal to the host cell when silenced. Three previous genome-wide RNAi experiments identified HDF sets with little overlap. We combine data from these three studies with a human protein interaction network to predict new HDFs, using an intuitive algorithm called SinkSource and four other algorithms published in the literature. Our algorithm achieves high precision and recall upon cross validation, as do the other methods. A number of HDFs that we predict are known to interact with HIV proteins. They belong to multiple protein complexes and biological processes that are known to be manipulated by HIV. We also demonstrate that many predicted HDF genes show significantly different programs of expression in early response to SIV infection in two non-human primate species that differ in AIDS progression. Our results suggest that many HDFs are yet to be discovered and that they have potential value as prognostic markers to determine pathological outcome and the likelihood of AIDS development. More generally, if multiple genome-wide gene-level studies have been performed at independent labs to study the same biological system or phenomenon, our methodology is applicable to interpret these studies simultaneously in the context of molecular interaction networks and to ask if they reinforce or contradict each other.
HIV 依赖因子(HDFs)是一类对 HIV 复制至关重要的人类蛋白,但当其被沉默时,对宿主细胞并无致死性。三项先前的全基因组 RNAi 实验鉴定出了重叠性很小的 HDF 集合。我们将这三项研究的数据与人类蛋白质相互作用网络相结合,使用一种名为 SinkSource 的直观算法和文献中发表的另外四种算法来预测新的 HDFs。我们的算法在交叉验证时具有高准确率和召回率,其他方法也是如此。我们预测的许多 HDFs已知与 HIV 蛋白相互作用。它们属于多个蛋白质复合物和生物过程,这些复合物和生物过程已知是被 HIV 操纵的。我们还证明,在两种在艾滋病进展方面存在差异的非人类灵长类动物中,许多预测的 HDF 基因在 SIV 感染后的早期反应中表现出显著不同的表达程序。我们的研究结果表明,许多 HDFs尚未被发现,它们具有作为预测标志物的潜在价值,可以确定病理结果和艾滋病发展的可能性。更一般地,如果多个独立实验室已经进行了全基因组基因水平的研究来研究相同的生物系统或现象,那么我们的方法适用于同时在分子相互作用网络的背景下解释这些研究,并询问它们是否相互支持或相互矛盾。