Midwest Respiratory Virus Program, Milwaukee, Wisconsin, United States of America.
PLoS One. 2011;6(10):e25468. doi: 10.1371/journal.pone.0025468. Epub 2011 Oct 6.
Respiratory Syncytial Virus (RSV) is the leading cause of lower respiratory-tract infections in infants and young children worldwide. Despite this, only six complete genome sequences of original strains have been previously published, the most recent of which dates back 35 and 26 years for RSV group A and group B respectively.
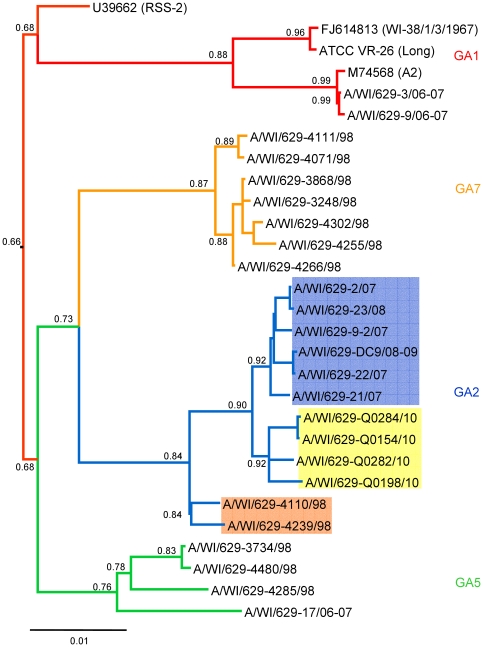
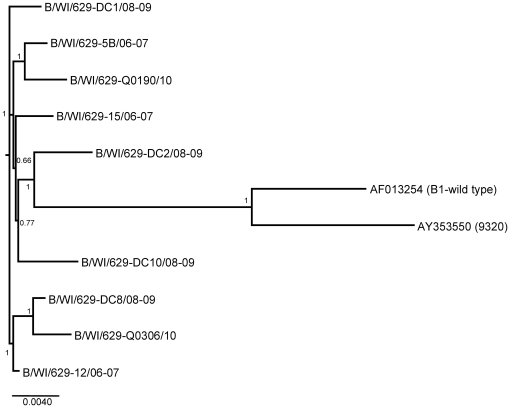
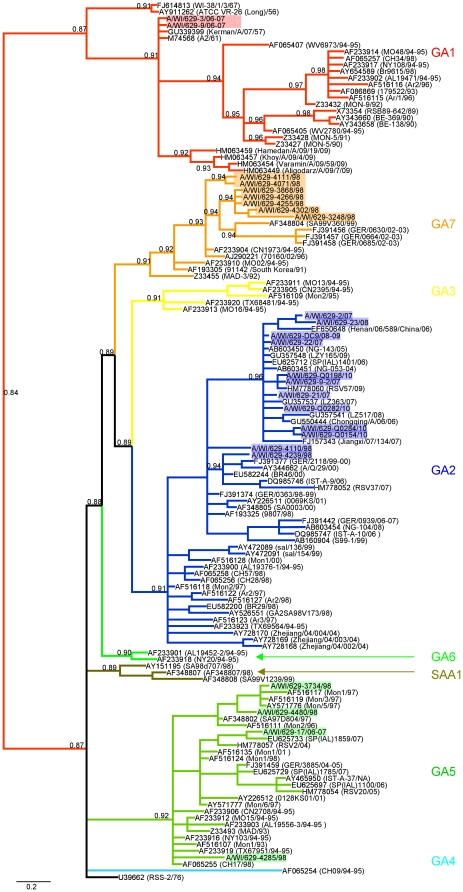
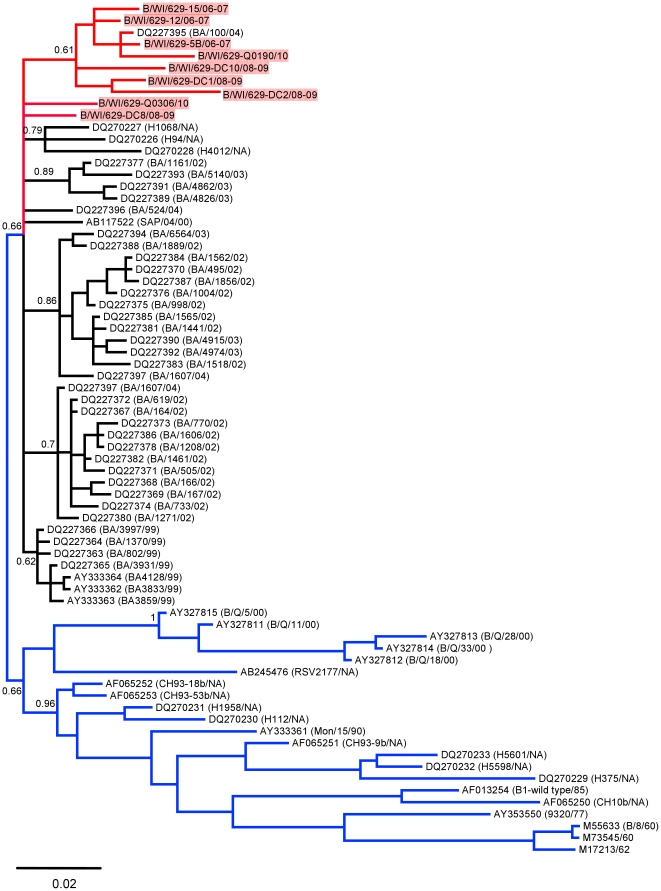
METHODOLOGY/PRINCIPAL FINDINGS: We present a semi-automated sequencing method allowing for the sequencing of four RSV whole genomes simultaneously. We were able to sequence the complete coding sequences of 13 RSV A and 4 RSV B strains from Milwaukee collected from 1998-2010. Another 12 RSV A and 5 RSV B strains sequenced in this study cover the majority of the genome. All RSV A and RSV B sequences were analyzed by neighbor-joining, maximum parsimony and Bayesian phylogeny methods. Genetic diversity was high among RSV A viruses in Milwaukee including the circulation of multiple genotypes (GA1, GA2, GA5, GA7) with GA2 persisting throughout the 13 years of the study. However, RSV B genomes showed little variation with all belonging to the BA genotype. For RSV A, the same evolutionary patterns and clades were seen consistently across the whole genome including all intergenic, coding, and non-coding regions sequences.
CONCLUSIONS/SIGNIFICANCE: The sequencing strategy presented in this work allows for RSV A and B genomes to be sequenced simultaneously in two working days and with a low cost. We have significantly increased the amount of genomic data that is available for both RSV A and B, providing the basic molecular characteristics of RSV strains circulating in Milwaukee over the last 13 years. This information can be used for comparative analysis with strains circulating in other communities around the world which should also help with the development of new strategies for control of RSV, specifically vaccine development and improvement of RSV diagnostics.
呼吸道合胞病毒(RSV)是全球婴幼儿下呼吸道感染的主要原因。尽管如此,之前仅发表了六株原始株的完整基因组序列,其中 RSV A 组和 RSV B 组的最新序列分别可追溯到 35 年和 26 年前。
方法/主要发现:我们提出了一种半自动测序方法,可同时对 4 株 RSV 全基因组进行测序。我们能够对从 1998 年至 2010 年在密尔沃基收集的 13 株 RSV A 和 4 株 RSV B 株进行完整编码序列测序。在本研究中测序的另外 12 株 RSV A 和 5 株 RSV B 株涵盖了大部分基因组。对所有 RSV A 和 RSV B 序列进行了邻接法、最大简约法和贝叶斯系统发生分析。密尔沃基 RSV A 病毒的遗传多样性很高,包括多种基因型(GA1、GA2、GA5、GA7)的循环,其中 GA2 在研究的 13 年中持续存在。然而,RSV B 基因组的变异很小,均属于 BA 基因型。对于 RSV A,整个基因组包括所有内含子、编码和非编码区序列,都始终存在相同的进化模式和分支。
结论/意义:本工作中提出的测序策略可在两个工作日内以低成本同时对 RSV A 和 B 基因组进行测序。我们大大增加了 RSV A 和 B 可用基因组数据的数量,提供了过去 13 年在密尔沃基循环的 RSV 株的基本分子特征。这些信息可用于与世界各地其他社区循环的菌株进行比较分析,这也有助于制定控制 RSV 的新策略,特别是疫苗开发和 RSV 诊断的改进。