Department of Pediatrics, Division of Metabolic Diseases and Genetics, Center for Lysosomal and Metabolic Diseases, Erasmus MC University Medical Center, PO Box 2060, 3000 CB Rotterdam, the Netherlands.
J Inherit Metab Dis. 2012 May;35(3):505-11. doi: 10.1007/s10545-011-9404-7. Epub 2011 Oct 19.
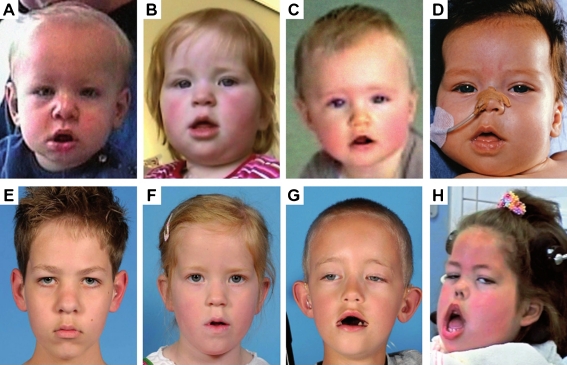
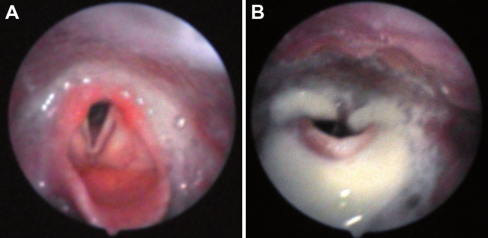
Classic infantile Pompe disease is an inherited generalized glycogen storage disorder caused by deficiency of lysosomal acid α-glucosidase. If left untreated, patients die before one year of age. Although enzyme-replacement therapy (ERT) has significantly prolonged lifespan, it has also revealed new aspects of the disease. For up to 11 years, we investigated the frequency and consequences of facial-muscle weakness, speech disorders and dysphagia in long-term survivors. Sequential photographs were used to determine the timing and severity of facial-muscle weakness. Using standardized articulation tests and fibreoptic endoscopic evaluation of swallowing, we investigated speech and swallowing function in a subset of patients. This study included 11 patients with classic infantile Pompe disease. Median age at the start of ERT was 2.4 months (range 0.1-8.3 months), and median age at the end of the study was 4.3 years (range 7.7 months -12.2 years). All patients developed facial-muscle weakness before the age of 15 months. Speech was studied in four patients. Articulation was disordered, with hypernasal resonance and reduced speech intelligibility in all four. Swallowing function was studied in six patients, the most important findings being ineffective swallowing with residues of food (5/6), penetration or aspiration (3/6), and reduced pharyngeal and/or laryngeal sensibility (2/6). We conclude that facial-muscle weakness, speech disorders and dysphagia are common in long-term survivors receiving ERT for classic infantile Pompe disease. To improve speech and reduce the risk for aspiration, early treatment by a speech therapist and regular swallowing assessments are recommended.
经典婴儿型庞贝病是一种遗传性的全身性糖原贮积病,由溶酶体酸性α-葡萄糖苷酶缺乏引起。如果不治疗,患者会在一岁前死亡。尽管酶替代疗法(ERT)显著延长了患者的寿命,但也揭示了该疾病的新方面。在长达 11 年的时间里,我们调查了长期存活者中面肌无力、言语障碍和吞咽困难的频率和后果。我们使用连续的照片来确定面肌无力的时间和严重程度。通过使用标准化的发音测试和纤维内镜吞咽评估,我们调查了部分患者的言语和吞咽功能。这项研究包括 11 名经典婴儿型庞贝病患者。ERT 开始时的中位年龄为 2.4 个月(范围为 0.1-8.3 个月),研究结束时的中位年龄为 4.3 岁(范围为 7.7 个月-12.2 岁)。所有患者在 15 个月前都出现了面肌无力。我们对四名患者进行了言语研究。所有患者的发音都出现了障碍,鼻音过重,言语清晰度降低。我们对六名患者进行了吞咽功能研究,最重要的发现是吞咽无效,食物残留(5/6),有渗漏或吸入(3/6),咽部和/或喉部感觉减退(2/6)。我们的结论是,接受经典婴儿型庞贝病 ERT 治疗的长期存活者中,面肌无力、言语障碍和吞咽困难很常见。为了改善言语功能并降低吸入风险,建议早期由言语治疗师进行治疗,并定期进行吞咽评估。