Brito Glauber C, Andrews David W
Department of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, Ontario L8N 3Z5, Canada.
BMC Syst Biol. 2011 Oct 19;5:169. doi: 10.1186/1752-0509-5-169.
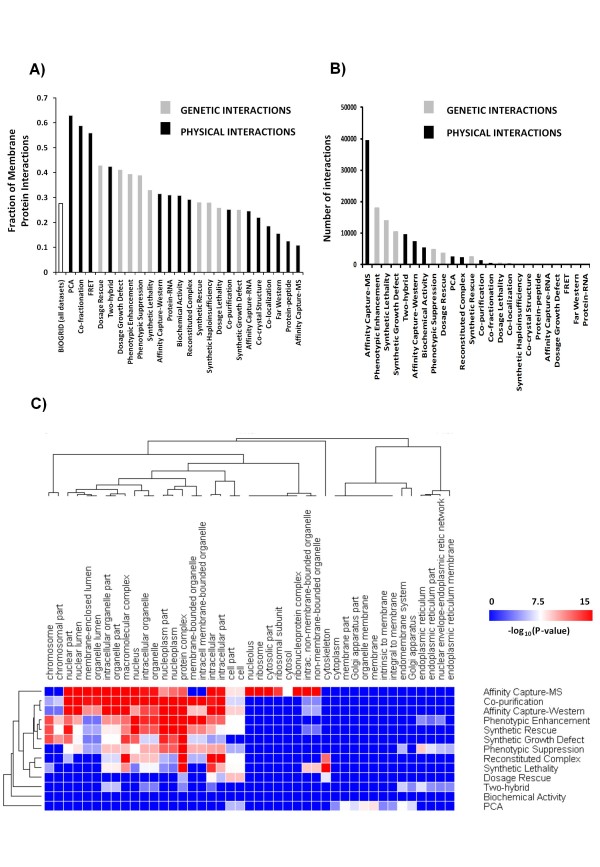
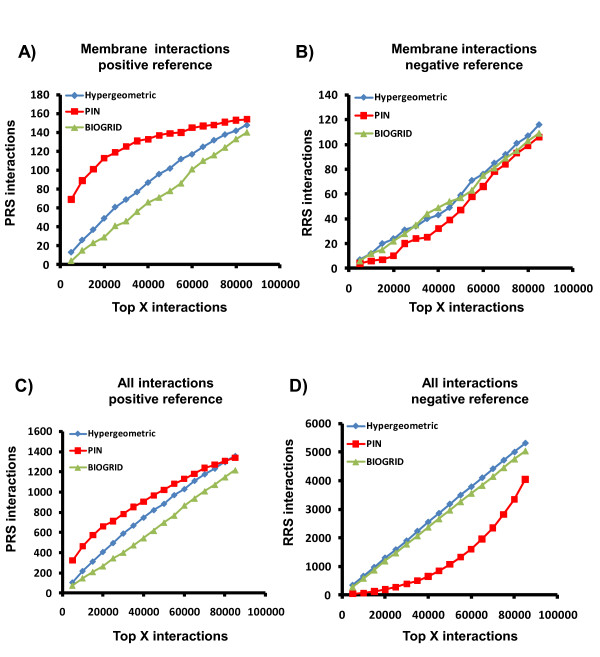
Cellular interaction networks can be used to analyze the effects on cell signaling and other functional consequences of perturbations to cellular physiology. Thus, several methods have been used to reconstitute interaction networks from multiple published datasets. However, the structure and performance of these networks depends on both the quality and the unbiased nature of the original data. Due to the inherent bias against membrane proteins in protein-protein interaction (PPI) data, interaction networks can be compromised particularly if they are to be used in conjunction with drug screening efforts, since most drug-targets are membrane proteins.
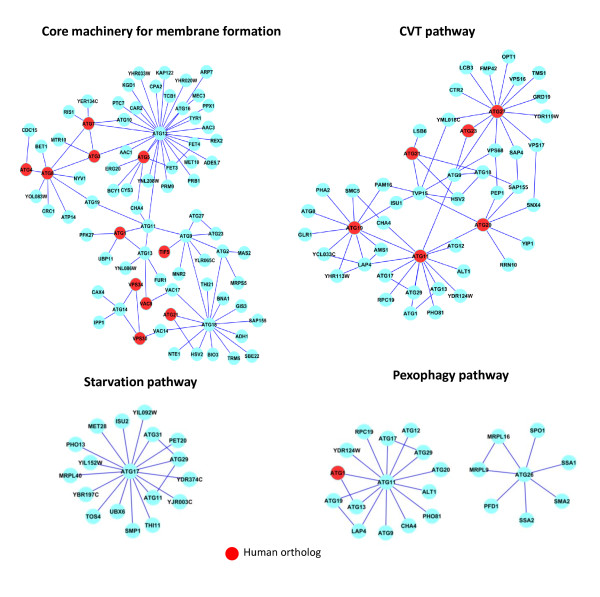
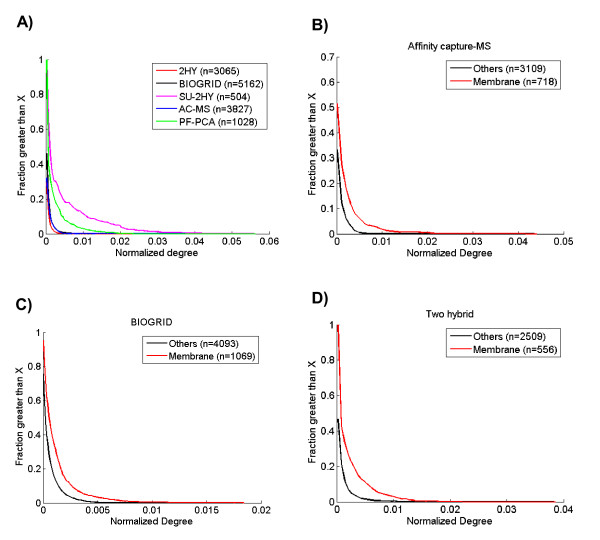
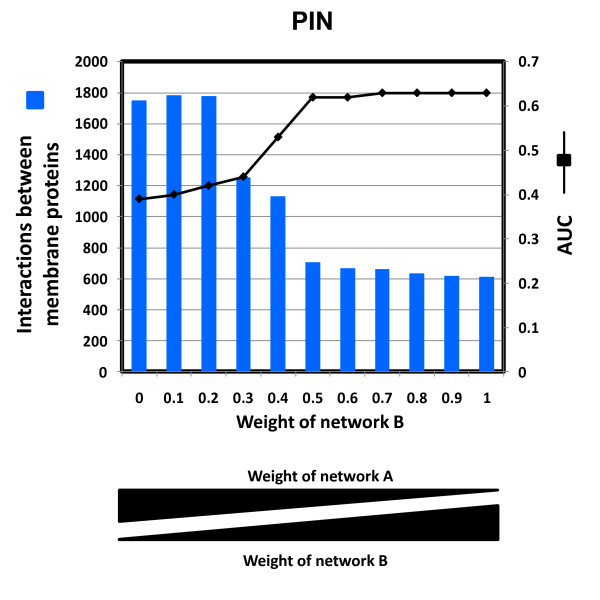
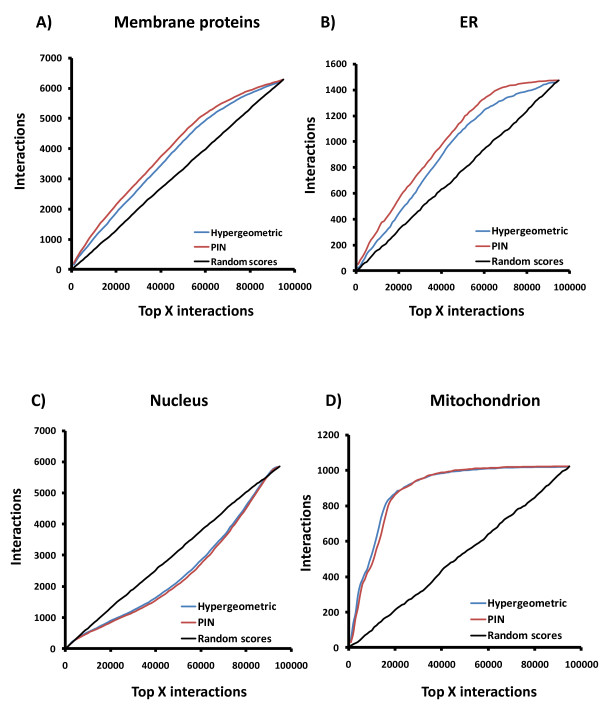
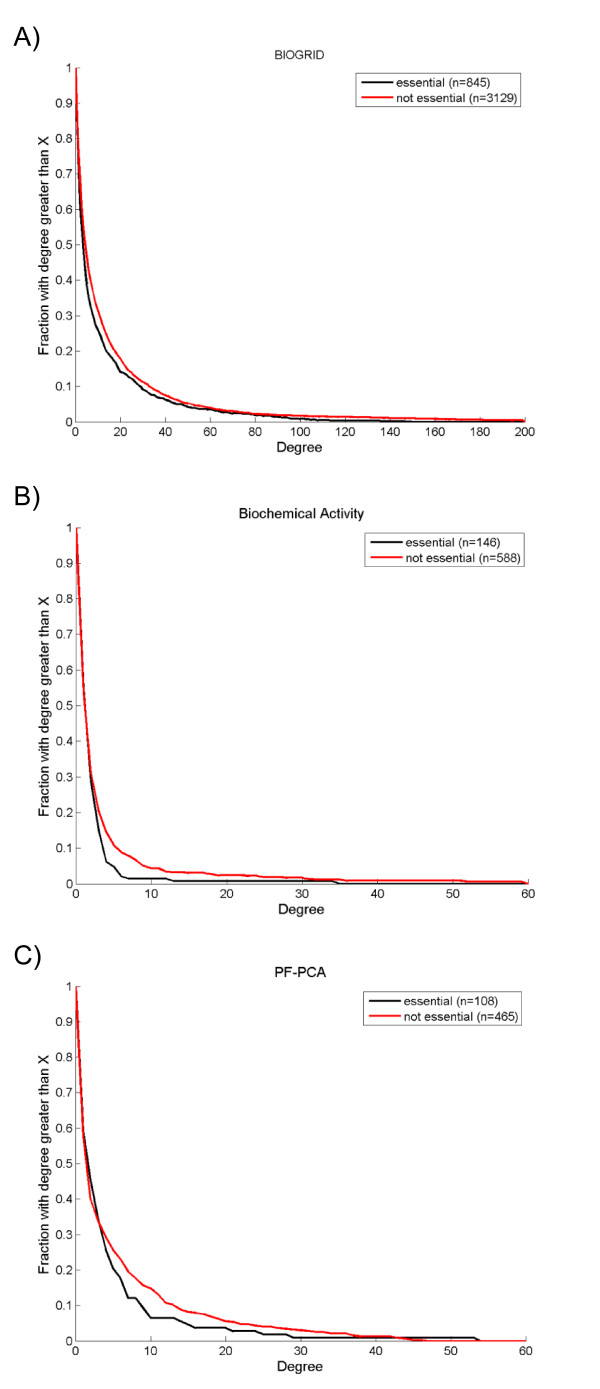
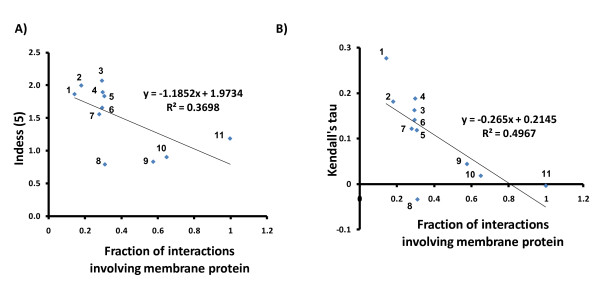
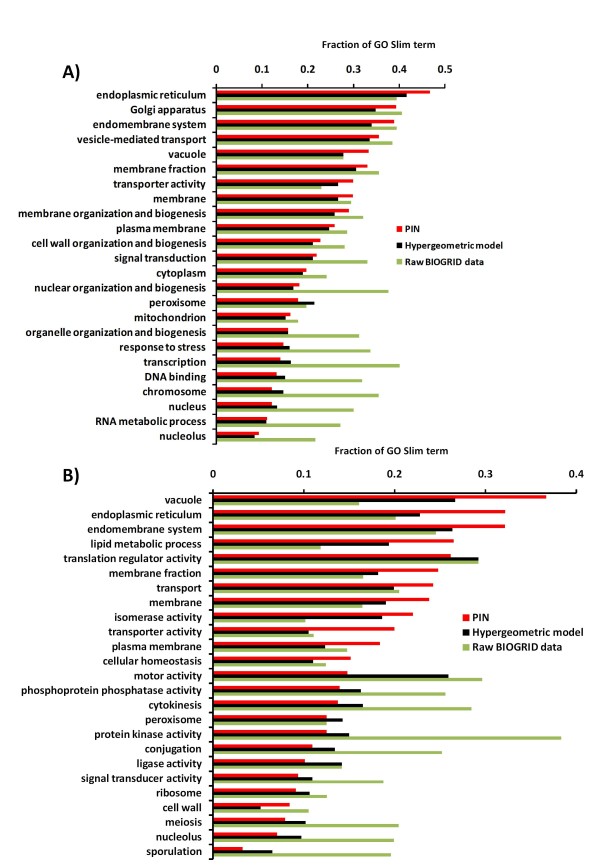
To overcome the experimental bias against PPIs involving membrane-associated proteins we used a probabilistic approach based on a hypergeometric distribution followed by logistic regression to simultaneously optimize the weights of different sources of interaction data. The resulting less biased genome-scale network constructed for the budding yeast Saccharomyces cerevisiae revealed that the starvation pathway is a distinct subnetwork of autophagy and retrieved a more integrated network of unfolded protein response genes. We also observed that the centrality-lethality rule depends on the content of membrane proteins in networks.
We show here that the bias against membrane proteins can and should be corrected in order to have a better representation of the interactions and topological properties of protein interaction networks.
细胞相互作用网络可用于分析细胞生理扰动对细胞信号传导及其他功能后果的影响。因此,已采用多种方法从多个已发表的数据集中重构相互作用网络。然而,这些网络的结构和性能取决于原始数据的质量和无偏性。由于蛋白质 - 蛋白质相互作用(PPI)数据中对膜蛋白存在固有偏差,相互作用网络可能会受到影响,特别是当它们与药物筛选工作结合使用时,因为大多数药物靶点都是膜蛋白。
为了克服针对涉及膜相关蛋白的PPI的实验偏差,我们使用了一种基于超几何分布的概率方法,随后进行逻辑回归,以同时优化不同相互作用数据源的权重。为芽殖酵母酿酒酵母构建的偏差较小的基因组规模网络表明,饥饿途径是自噬的一个独特子网,并检索到一个更完整的未折叠蛋白反应基因网络。我们还观察到中心性 - 致死性规则取决于网络中膜蛋白的含量。
我们在此表明,针对膜蛋白的偏差能够且应该得到纠正,以便更好地呈现蛋白质相互作用网络的相互作用和拓扑特性。