Stanford Genome Technology Center, Stanford University, Palo Alto, CA 94304, USA.
Nucleic Acids Res. 2012 Jan;40(1):e2. doi: 10.1093/nar/gkr861. Epub 2011 Oct 19.
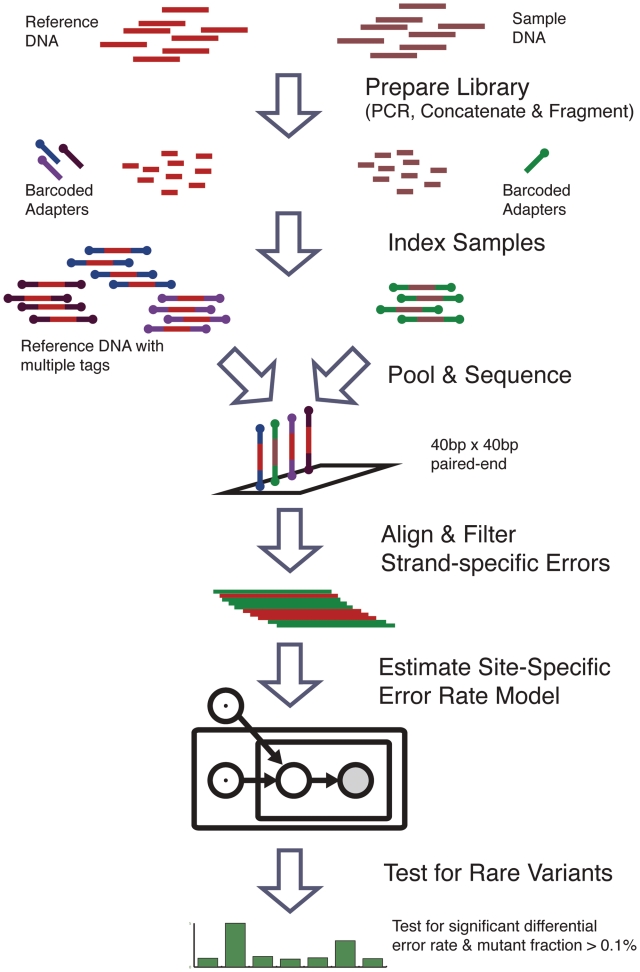
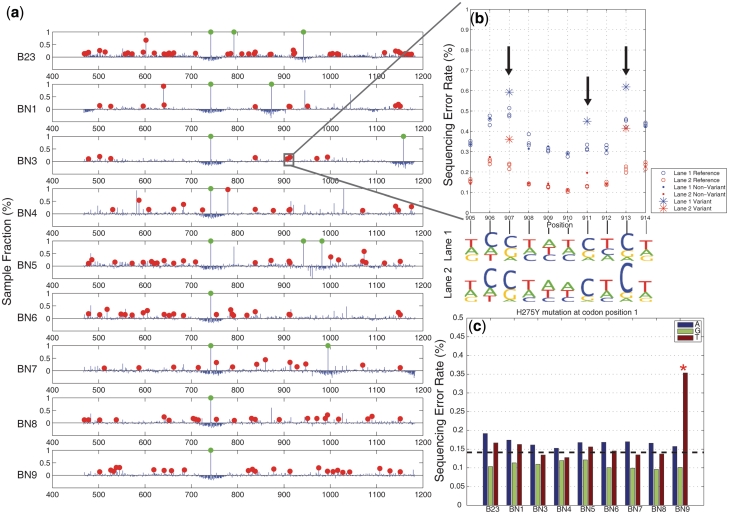
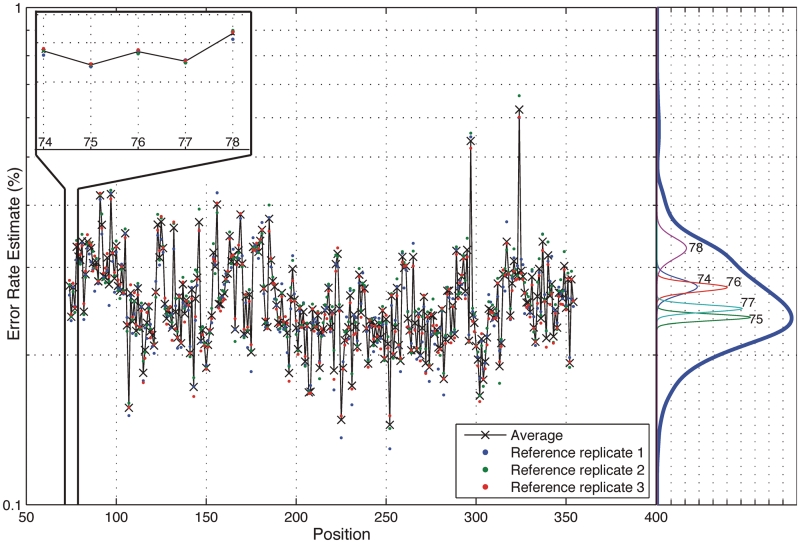
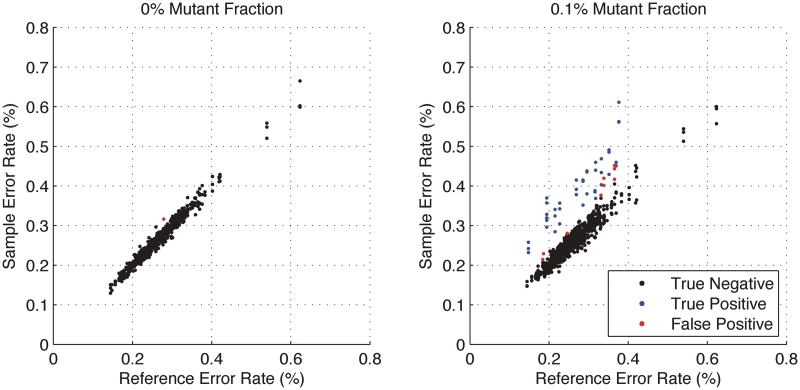
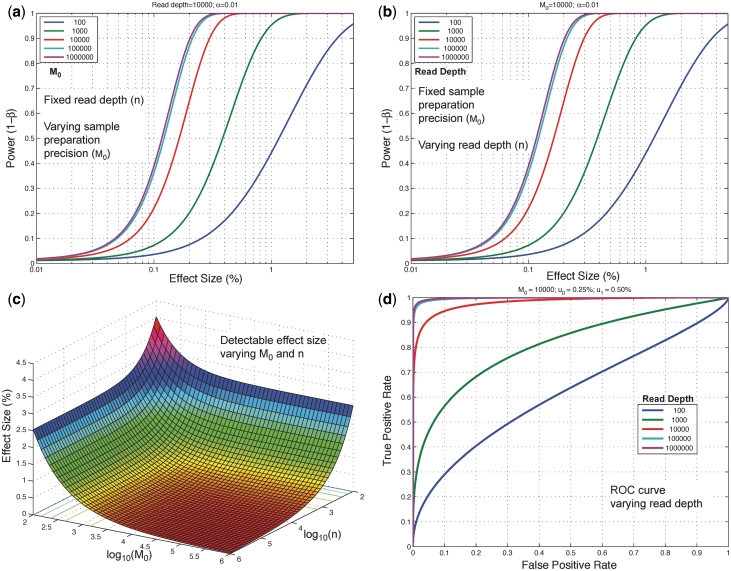
With next-generation DNA sequencing technologies, one can interrogate a specific genomic region of interest at very high depth of coverage and identify less prevalent, rare mutations in heterogeneous clinical samples. However, the mutation detection levels are limited by the error rate of the sequencing technology as well as by the availability of variant-calling algorithms with high statistical power and low false positive rates. We demonstrate that we can robustly detect mutations at 0.1% fractional representation. This represents accurate detection of one mutant per every 1000 wild-type alleles. To achieve this sensitive level of mutation detection, we integrate a high accuracy indexing strategy and reference replication for estimating sequencing error variance. We employ a statistical model to estimate the error rate at each position of the reference and to quantify the fraction of variant base in the sample. Our method is highly specific (99%) and sensitive (100%) when applied to a known 0.1% sample fraction admixture of two synthetic DNA samples to validate our method. As a clinical application of this method, we analyzed nine clinical samples of H1N1 influenza A and detected an oseltamivir (antiviral therapy) resistance mutation in the H1N1 neuraminidase gene at a sample fraction of 0.18%.
利用下一代 DNA 测序技术,人们可以在非常高的覆盖深度下对特定的基因组感兴趣区域进行检测,并在异质临床样本中识别出不太常见的罕见突变。然而,突变检测水平受到测序技术的错误率以及具有高统计能力和低假阳性率的变异调用算法的可用性的限制。我们证明,我们可以稳健地检测到 0.1%分数代表的突变。这代表着每 1000 个野生型等位基因中准确检测到一个突变。为了实现这种敏感的突变检测水平,我们整合了一种高精度索引策略和参考复制,以估计测序误差方差。我们采用一种统计模型来估计参考中每个位置的错误率,并量化样本中变异碱基的分数。当应用于验证我们方法的两个合成 DNA 样本的已知 0.1%样本分数混合物时,我们的方法具有高度的特异性(99%)和敏感性(100%)。作为该方法的临床应用,我们分析了 9 个甲型 H1N1 流感临床样本,并在样本分数为 0.18%时检测到 H1N1 神经氨酸酶基因中的奥司他韦(抗病毒治疗)耐药突变。