Department of Pathology, The University of Michigan Medical School, Ann Arbor, Michigan, United States of America.
PLoS Genet. 2011 Oct;7(10):e1002321. doi: 10.1371/journal.pgen.1002321. Epub 2011 Oct 13.
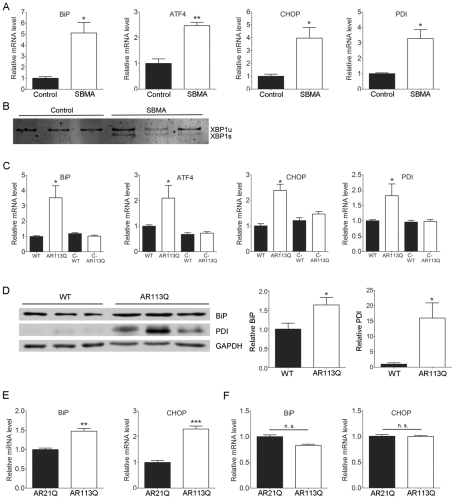
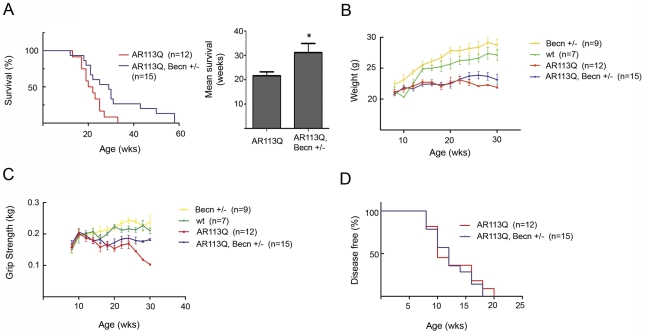
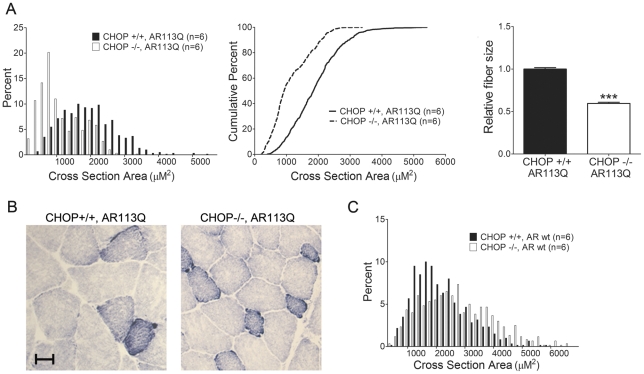
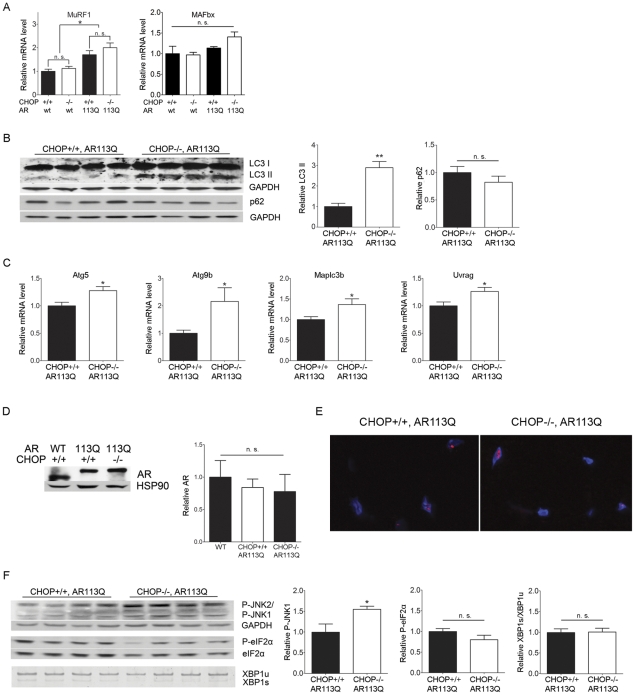
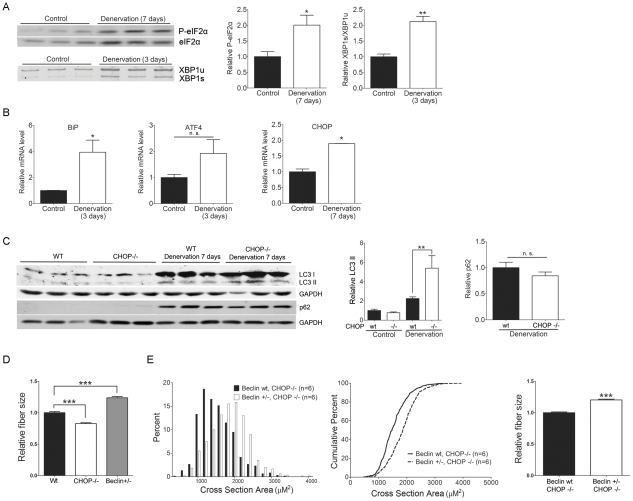
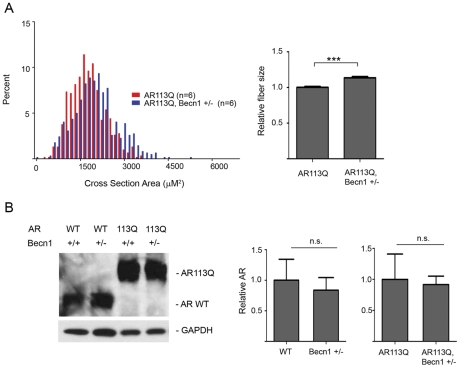
Altered protein homeostasis underlies degenerative diseases triggered by misfolded proteins, including spinal and bulbar muscular atrophy (SBMA), a neuromuscular disorder caused by a CAG/glutamine expansion in the androgen receptor. Here we show that the unfolded protein response (UPR), an ER protein quality control pathway, is induced in skeletal muscle from SBMA patients, AR113Q knock-in male mice, and surgically denervated wild-type mice. To probe the consequence of UPR induction, we deleted CHOP (C/EBP homologous protein), a transcription factor induced following ER stress. CHOP deficiency accentuated atrophy in both AR113Q and surgically denervated muscle through activation of macroautophagy, a lysosomal protein quality control pathway. Conversely, impaired autophagy due to Beclin-1 haploinsufficiency decreased muscle wasting and extended lifespan of AR113Q males, producing a significant and unexpected amelioration of the disease phenotype. Our findings highlight critical cross-talk between the UPR and macroautophagy, and they indicate that autophagy activation accentuates aspects of the SBMA phenotype.
错误折叠的蛋白质引发的退行性疾病的基础是蛋白质稳态的改变,包括脊髓和延髓肌肉萎缩症(SBMA),这是一种由雄激素受体中的 CAG/谷氨酰胺扩展引起的神经肌肉疾病。在这里,我们表明,未折叠蛋白反应(UPR),一种内质网蛋白质量控制途径,在 SBMA 患者、AR113Q 基因敲入雄性小鼠和手术去神经的野生型小鼠的骨骼肌中被诱导。为了探究 UPR 诱导的后果,我们敲除了 CHOP(C/EBP 同源蛋白),这是一种在内质网应激后诱导的转录因子。CHOP 缺陷通过激活溶酶体蛋白质量控制途径巨自噬,加重了 AR113Q 和手术去神经肌肉的萎缩。相反,由于 Beclin-1 杂合不足导致的自噬受损,减少了肌肉消耗并延长了 AR113Q 雄性的寿命,显著改善了疾病表型。我们的发现强调了 UPR 和巨自噬之间的关键串扰,并表明自噬的激活加重了 SBMA 表型的某些方面。