Khan Arif O, Aldahmesh Mohammed A, Alkuraya Fowzan S
Division of Pediatric Ophthalmology, King Khaled Eye Specialist Hospital, Riyadh, Saudi Arabia.
Mol Vis. 2011;17:2570-9. Epub 2011 Oct 4.
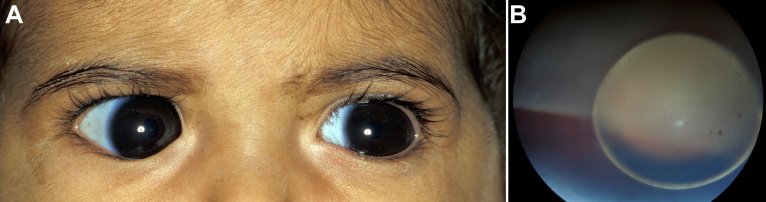
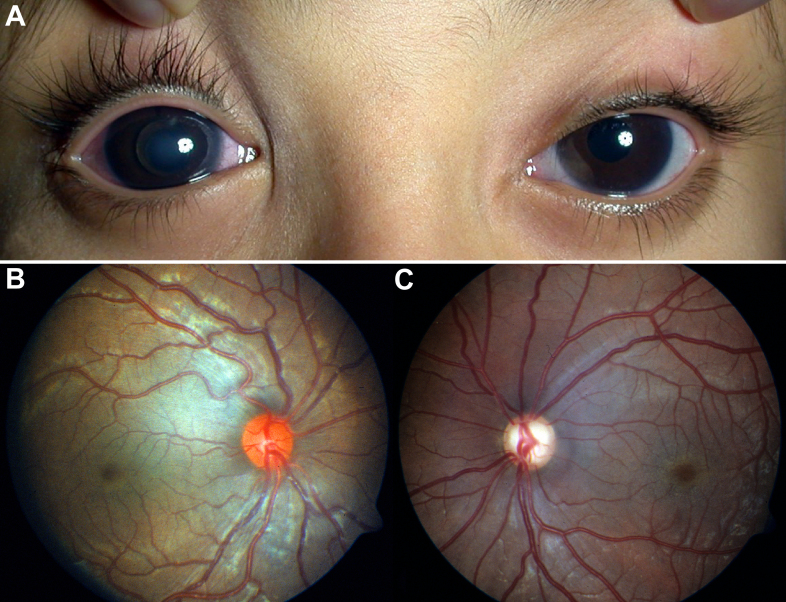
To clinically and genetically characterize a distinct phenotype of congenital megalocornea (horizontal corneal diameter ≥13 mm) with secondary glaucoma from spherophakia and/or ectopia lentis during childhood in affected Saudi families.
Clinical exam, homozygosity scan, and candidate gene analysis.
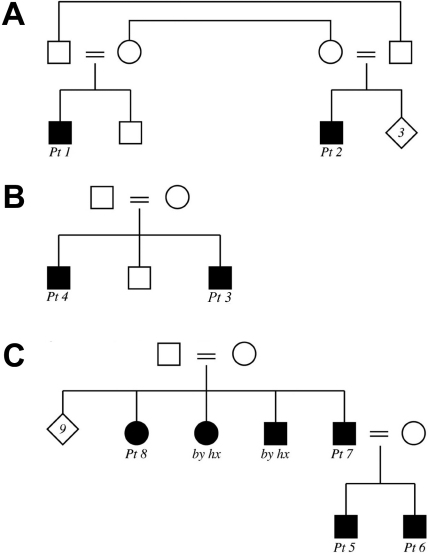
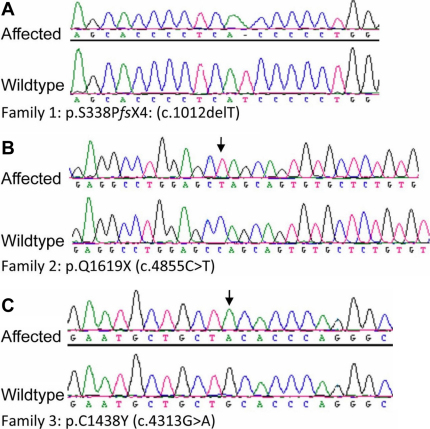
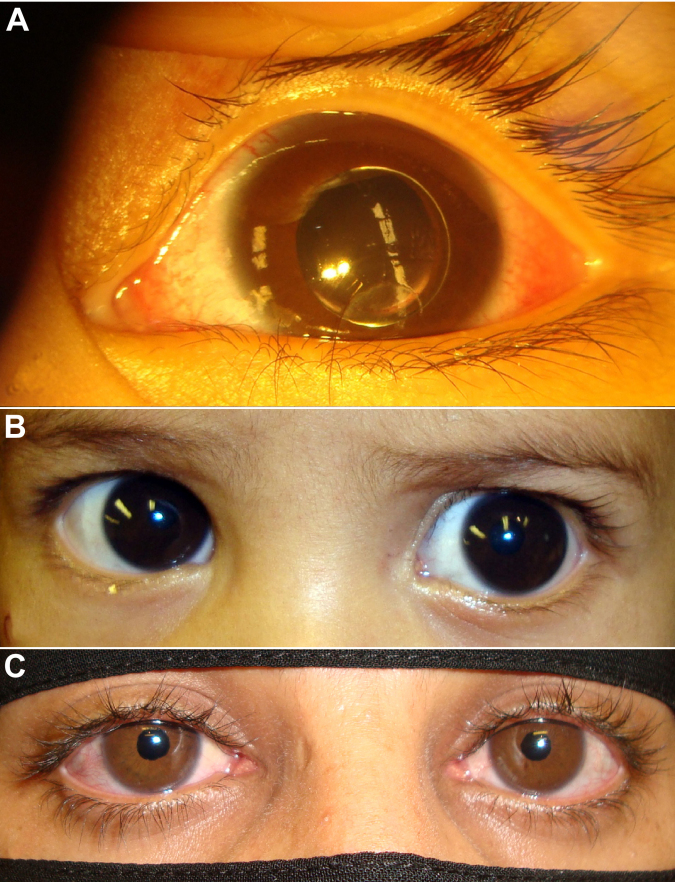
From 2005 to 2010, eight affected individuals from three consanguineous families were identified. In addition to congenital megalocornea, affected children presented with secondary glaucoma from spherophakia and/or ectopia lentis. One member from each family developed spontaneous complete crystalline lens dislocation into the anterior chamber with associated acute glaucoma during early childhood. Older individuals had phenotypes that would have suggested prior uncontrolled primary congenital/infantile glaucoma had past ophthalmic and/or family histories not been available. Homozygosity mapping performed for the first two families suggested the candidate gene latent transforming growth factor-beta-binding protein 2 (LTBP2), which when sequenced revealed a novel homozgyous mutation that segregated with the phenotype in each family (p.S338PfsX4 [c.1012delT], p.Q1619X[(c.4855C>T]). LTBP2 sequencing in the third family revealed a third novel homozygous mutation (p.C1438Y [c.4313G>A]).
Congenital megalocornea with childhood secondary glaucoma from spherophakia and/or ectopia lentis is a distinct condition caused by recessive LTBP2 mutations that needs to be distinguished from buphthalmos secondary to primary congenital/infantile glaucoma because typical initial surgical treatment is lens removal in the former and angle surgery in the latter. Complete dislocation of the crystalline lens into the anterior chamber during early childhood can occur in young children with this unique phenotype.
对沙特受影响家庭中患有先天性巨角膜(水平角膜直径≥13毫米)并伴有因球形晶状体和/或晶状体异位导致的继发性青光眼的独特表型进行临床和基因特征分析。
临床检查、纯合性扫描和候选基因分析。
2005年至2010年,共识别出三个近亲家庭中的八名受影响个体。除先天性巨角膜外,受影响儿童还患有因球形晶状体和/或晶状体异位导致的继发性青光眼。每个家庭中有一名成员在幼儿期出现晶状体自发完全脱入前房并伴有急性青光眼。如果没有既往眼科和/或家族病史,年龄较大的个体所具有的表型可能提示曾患有未经控制的原发性先天性/婴儿性青光眼。对前两个家庭进行的纯合性定位分析提示候选基因潜在转化生长因子β结合蛋白2(LTBP2),对其进行测序后发现一个新的纯合突变,该突变在每个家庭中均与表型共分离(p.S338PfsX4 [c.1012delT],p.Q1619X[(c.4855C>T])。对第三个家庭进行的LTBP2测序发现了第三个新的纯合突变(p.C1438Y [c.4313G>A])。
先天性巨角膜伴有因球形晶状体和/或晶状体异位导致的儿童继发性青光眼是一种由隐性LTBP2突变引起的独特病症,需要与原发性先天性/婴儿性青光眼继发的牛眼症相区分,因为前者典型的初始手术治疗是晶状体摘除,而后者是房角手术。患有这种独特表型的幼儿在幼儿期可能出现晶状体完全脱入前房的情况。