Department of Biology, East Carolina University, Greenville, North Carolina, United States of America.
PLoS One. 2011;6(11):e26980. doi: 10.1371/journal.pone.0026980. Epub 2011 Nov 8.
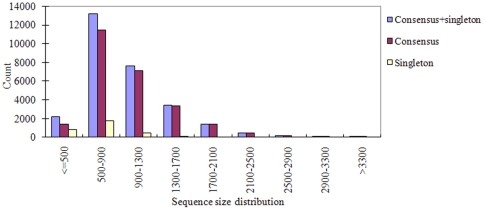
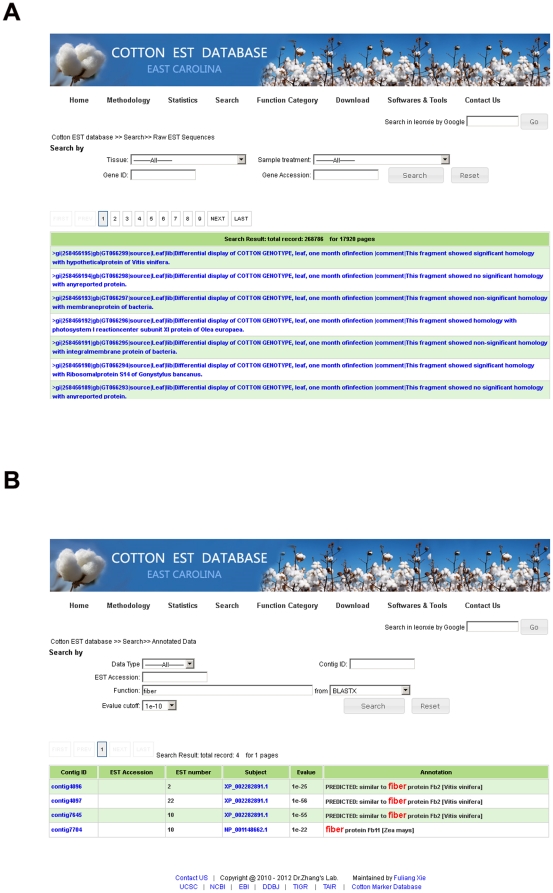
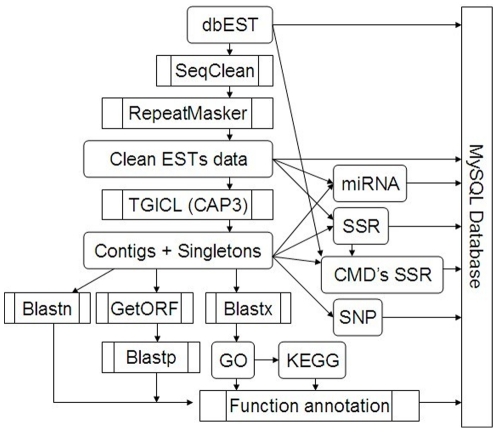
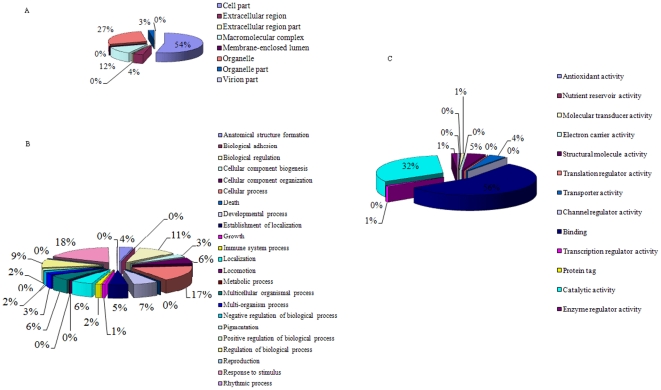
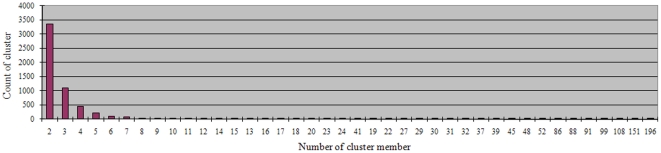
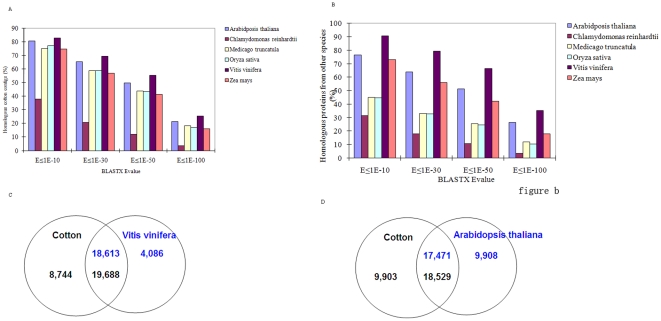
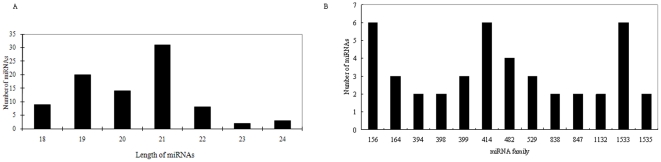
A total of 28,432 unique contigs (25,371 in consensus contigs and 3,061 as singletons) were assembled from all 268,786 cotton ESTs currently available. Several in silico approaches [comparative genomics, Blast, Gene Ontology (GO) analysis, and pathway enrichment by Kyoto Encyclopedia of Genes and Genomes (KEGG)] were employed to investigate global functions of the cotton transcriptome. Cotton EST contigs were clustered into 5,461 groups with a maximum cluster size of 196 members. A total of 27,956 indel mutants and 149,616 single nucleotide polymorphisms (SNPs) were identified from consensus contigs. Interestingly, many contigs with significantly high frequencies of indels or SNPs encode transcription factors and protein kinases. In a comparison with six model plant species, cotton ESTs show the highest overall similarity to grape. A total of 87 cotton miRNAs were identified; 59 of these have not been reported previously from experimental or bioinformatics investigations. We also predicted 3,260 genes as miRNAs targets, which are associated with multiple biological functions, including stress response, metabolism, hormone signal transduction and fiber development. We identified 151 and 4,214 EST-simple sequence repeats (SSRs) from contigs and raw ESTs respectively. To make these data widely available, and to facilitate access to EST-related genetic information, we integrated our results into a comprehensive, fully downloadable web-based cotton EST database (www.leonxie.com).
从目前所有 268786 个棉花 EST 中总共组装了 28432 个独特的连续体(25371 个在共识连续体中,3061 个作为单一体)。采用了几种计算机方法[比较基因组学、Blast、基因本体论(GO)分析和京都基因与基因组百科全书(KEGG)的途径富集]来研究棉花转录组的全局功能。棉花 EST 连续体分为 5461 组,最大聚类大小为 196 个成员。从共识连续体中总共鉴定出 27956 个插入缺失突变和 149616 个单核苷酸多态性(SNP)。有趣的是,许多具有高频率插入缺失或 SNP 的连续体编码转录因子和蛋白激酶。与六个模式植物物种相比,棉花 EST 与葡萄的总体相似性最高。总共鉴定出 87 个棉花 miRNA,其中 59 个以前没有通过实验或生物信息学研究报道过。我们还预测了 3260 个作为 miRNA 靶标的基因,这些基因与多种生物学功能有关,包括应激反应、代谢、激素信号转导和纤维发育。我们分别从连续体和原始 EST 中鉴定出 151 个和 4214 个 EST-简单序列重复(SSR)。为了广泛提供这些数据,并方便访问 EST 相关遗传信息,我们将我们的结果整合到一个全面的、可完全下载的基于网络的棉花 EST 数据库(www.leonxie.com)中。