Department of Biology, Institute of Molecular Systems Biology, ETH Zürich, 8093 Zürich, Switzerland.
Mol Cell Proteomics. 2012 Mar;11(3):M111.013987. doi: 10.1074/mcp.M111.013987. Epub 2011 Nov 20.
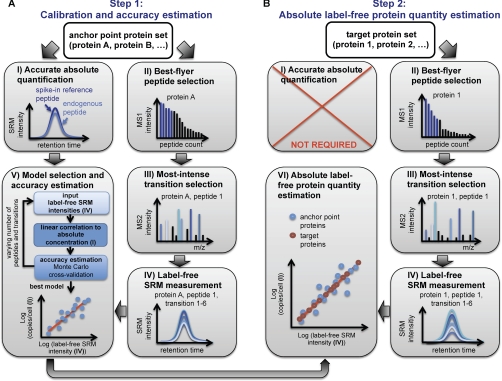
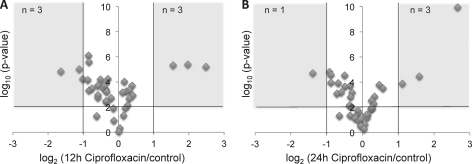
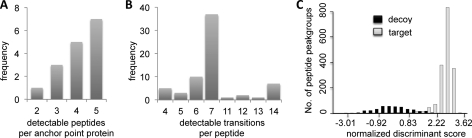
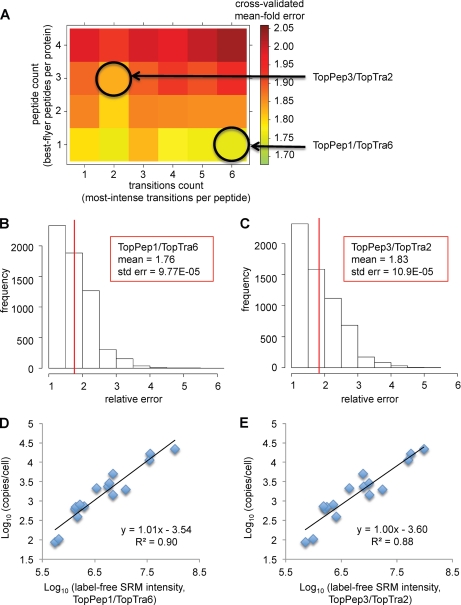
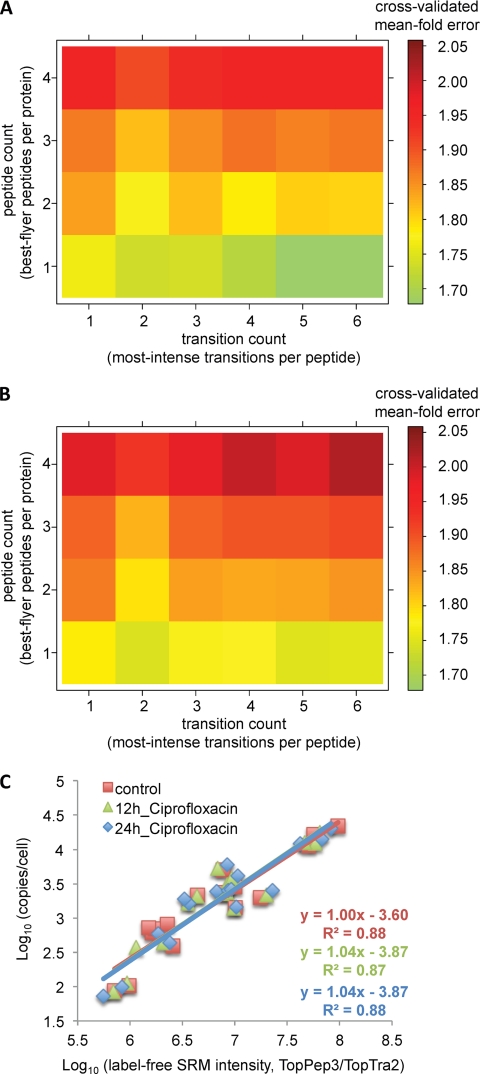
For many research questions in modern molecular and systems biology, information about absolute protein quantities is imperative. This information includes, for example, kinetic modeling of processes, protein turnover determinations, stoichiometric investigations of protein complexes, or quantitative comparisons of different proteins within one sample or across samples. To date, the vast majority of proteomic studies are limited to providing relative quantitative comparisons of protein levels between limited numbers of samples. Here we describe and demonstrate the utility of a targeting MS technique for the estimation of absolute protein abundance in unlabeled and nonfractionated cell lysates. The method is based on selected reaction monitoring (SRM) mass spectrometry and the "best flyer" hypothesis, which assumes that the specific MS signal intensity of the most intense tryptic peptides per protein is approximately constant throughout a whole proteome. SRM-targeted best flyer peptides were selected for each protein from the peptide precursor ion signal intensities from directed MS data. The most intense transitions per peptide were selected from full MS/MS scans of crude synthetic analogs. We used Monte Carlo cross-validation to systematically investigate the accuracy of the technique as a function of the number of measured best flyer peptides and the number of SRM transitions per peptide. We found that a linear model based on the two most intense transitions of the three best flying peptides per proteins (TopPep3/TopTra2) generated optimal results with a cross-correlated mean fold error of 1.8 and a squared Pearson coefficient R(2) of 0.88. Applying the optimized model to lysates of the microbe Leptospira interrogans, we detected significant protein abundance changes of 39 target proteins upon antibiotic treatment, which correlate well with literature values. The described method is generally applicable and exploits the inherent performance advantages of SRM, such as high sensitivity, selectivity, reproducibility, and dynamic range, and estimates absolute protein concentrations of selected proteins at minimized costs.
对于现代分子和系统生物学中的许多研究问题,绝对蛋白质数量的信息是必不可少的。这些信息包括,例如,过程的动力学建模、蛋白质周转率的测定、蛋白质复合物的化学计量研究,或在一个样品或多个样品中对不同蛋白质进行定量比较。迄今为止,绝大多数蛋白质组学研究仅限于提供有限数量的样品之间蛋白质水平的相对定量比较。在这里,我们描述并展示了一种靶向 MS 技术在未标记和未分级细胞裂解物中估计绝对蛋白质丰度的实用性。该方法基于选择反应监测 (SRM) 质谱和“最佳飞行”假设,该假设假定每个蛋白质的最强烈的胰蛋白酶肽的特定 MS 信号强度在整个蛋白质组中大致恒定。从定向 MS 数据的肽前体离子信号强度中为每个蛋白质选择 SRM 靶向最佳飞行肽。从粗合成类似物的全 MS/MS 扫描中选择每个肽的最强转换。我们使用蒙特卡罗交叉验证系统地研究了该技术作为测量的最佳飞行肽数量和每个肽的 SRM 转换数量的函数的准确性。我们发现,基于每个蛋白质的三个最佳飞行肽的两个最强转换的线性模型(TopPep3/TopTra2)生成了最佳结果,交叉相关平均折叠误差为 1.8,平方 Pearson 系数 R(2)为 0.88。将优化的模型应用于微生物钩端螺旋体的裂解物,我们检测到抗生素处理后 39 个靶蛋白的显著蛋白质丰度变化,与文献值很好地相关。所描述的方法具有普遍适用性,并利用了 SRM 的固有性能优势,例如高灵敏度、选择性、重现性和动态范围,并以最小的成本估计选定蛋白质的绝对蛋白质浓度。