Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, Connecticut 06536-8012, USA.
J Biol Chem. 2012 Jan 20;287(4):2558-67. doi: 10.1074/jbc.M111.316760. Epub 2011 Nov 28.
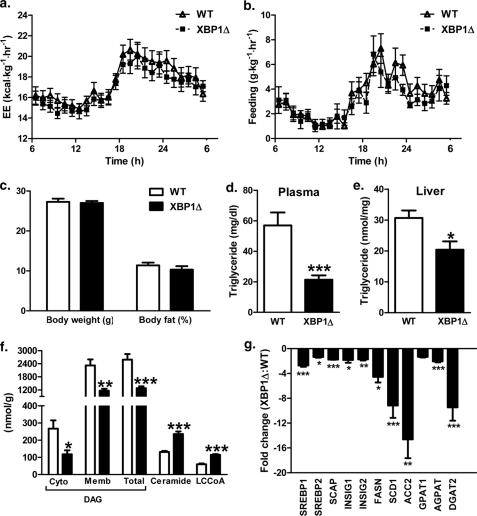
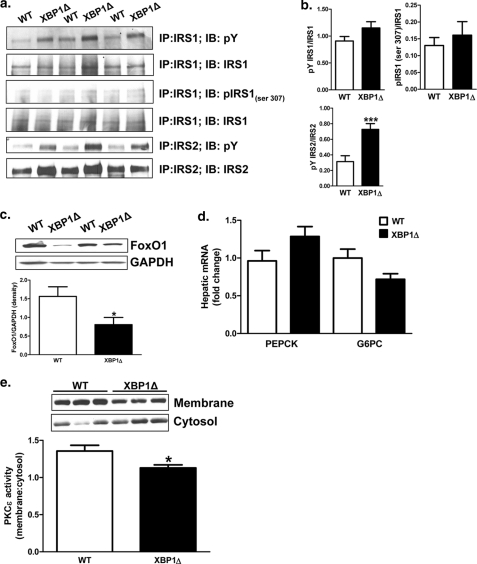
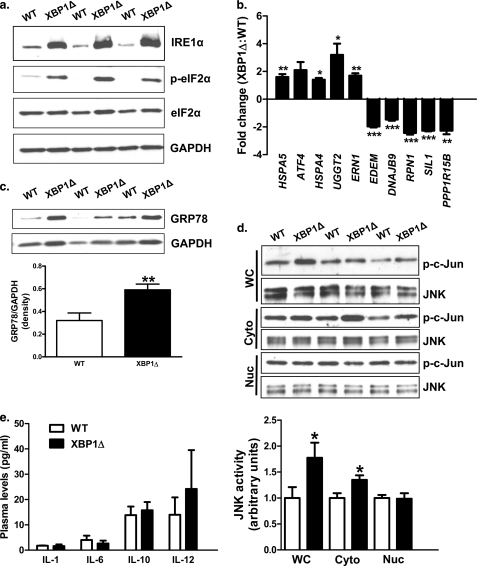
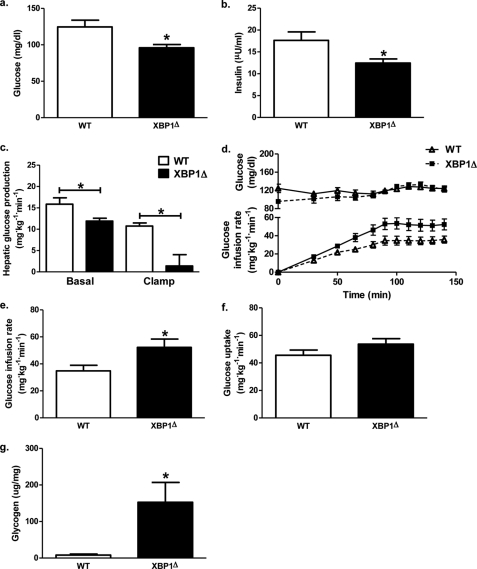
Hepatic insulin resistance has been attributed to both increased endoplasmic reticulum (ER) stress and accumulation of intracellular lipids, specifically diacylglycerol (DAG). The ER stress response protein, X-box-binding protein-1 (XBP1), was recently shown to regulate hepatic lipogenesis, suggesting that hepatic insulin resistance in models of ER stress may result from defective lipid storage, as opposed to ER-specific stress signals. Studies were designed to dissociate liver lipid accumulation and activation of ER stress signaling pathways, which would allow us to delineate the individual contributions of ER stress and hepatic lipid content to the pathogenesis of hepatic insulin resistance. Conditional XBP1 knock-out (XBP1Δ) and control mice were fed fructose chow for 1 week. Determinants of whole-body energy balance, weight, and composition were determined. Hepatic lipids including triglyceride, DAGs, and ceramide were measured, alongside markers of ER stress. Whole-body and tissue-specific insulin sensitivity were determined by hyperinsulinemic-euglycemic clamp studies. Hepatic ER stress signaling was increased in fructose chow-fed XBP1Δ mice as reflected by increased phosphorylated eIF2α, HSPA5 mRNA, and a 2-fold increase in hepatic JNK activity. Despite JNK activation, XBP1Δ displayed increased hepatic insulin sensitivity during hyperinsulinemic-euglycemic clamp studies, which was associated with increased insulin-stimulated IRS2 tyrosine phosphorylation, reduced hepatic DAG content, and reduced PKCε activity. These studies demonstrate that ER stress and IRE1α-mediated JNK activation can be disassociated from hepatic insulin resistance and support the hypothesis that hepatic insulin resistance in models of ER stress may be secondary to ER stress modulation of hepatic lipogenesis.
肝脏胰岛素抵抗归因于内质网(ER)应激的增加和细胞内脂质的积累,特别是二酰基甘油(DAG)。最近发现,内质网应激反应蛋白 X 盒结合蛋白-1(XBP1)调节肝脂肪生成,这表明 ER 应激模型中的肝脏胰岛素抵抗可能是由于脂质储存缺陷,而不是 ER 特异性应激信号。本研究旨在分离肝脏脂质积累和 ER 应激信号通路的激活,这将使我们能够描绘 ER 应激和肝脂质含量对肝脏胰岛素抵抗发病机制的各自贡献。条件性 XBP1 敲除(XBP1Δ)和对照小鼠用果糖饲料喂养 1 周。测定全身能量平衡、体重和组成的决定因素。测定肝脂质,包括甘油三酯、DAG 和神经酰胺,以及 ER 应激的标志物。通过高胰岛素-正葡萄糖钳夹研究测定全身和组织特异性胰岛素敏感性。果糖饲料喂养的 XBP1Δ 小鼠的肝脏 ER 应激信号增加,反映在磷酸化 eIF2α、HSPA5mRNA 增加和 JNK 活性增加 2 倍。尽管 JNK 激活,XBP1Δ 在高胰岛素-正葡萄糖钳夹研究中显示出增加的肝脏胰岛素敏感性,这与胰岛素刺激的 IRS2 酪氨酸磷酸化增加、肝 DAG 含量减少和 PKCε 活性降低有关。这些研究表明,内质网应激和 IRE1α 介导的 JNK 激活可以与肝脏胰岛素抵抗分离,并支持内质网应激模型中肝脏胰岛素抵抗可能继发于 ER 应激对肝脂肪生成的调节的假说。