Department of Computer Science, The University of Texas at San Antonio, One UTSA Circle, San Antonio, Texas 78249, USA.
BMC Bioinformatics. 2011 Nov 24;12 Suppl 12(Suppl 12):S2. doi: 10.1186/1471-2105-12-S12-S2.
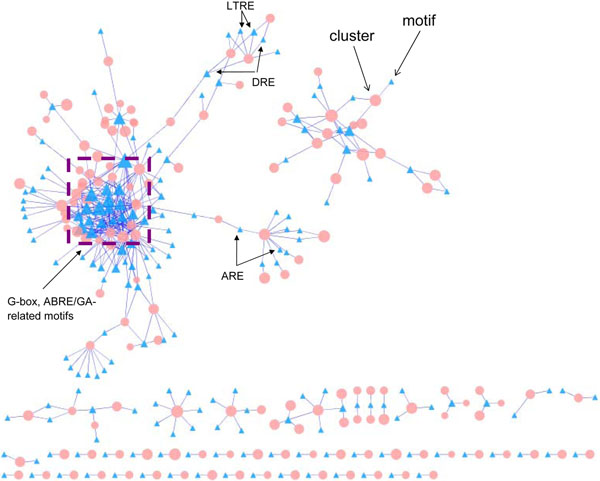
Several large-scale gene co-expression networks have been constructed successfully for predicting gene functional modules and cis-regulatory elements in Arabidopsis (Arabidopsis thaliana). However, these networks are usually constructed and analyzed in an ad hoc manner. In this study, we propose a completely parameter-free and systematic method for constructing gene co-expression networks and predicting functional modules as well as cis-regulatory elements.
Our novel method consists of an automated network construction algorithm, a parameter-free procedure to predict functional modules, and a strategy for finding known cis-regulatory elements that is suitable for consensus scanning without prior knowledge of the allowed extent of degeneracy of the motif. We apply the method to study a large collection of gene expression microarray data in Arabidopsis. We estimate that our co-expression network has ~94% of accuracy, and has topological properties similar to other biological networks, such as being scale-free and having a high clustering coefficient. Remarkably, among the ~300 predicted modules whose sizes are at least 20, 88% have at least one significantly enriched functions, including a few extremely significant ones (ribosome, p < 1E-300, photosynthetic membrane, p < 1.3E-137, proteasome complex, p < 5.9E-126). In addition, we are able to predict cis-regulatory elements for 66.7% of the modules, and the association between the enriched cis-regulatory elements and the enriched functional terms can often be confirmed by the literature. Overall, our results are much more significant than those reported by several previous studies on similar data sets. Finally, we utilize the co-expression network to dissect the promoters of 19 Arabidopsis genes involved in the metabolism and signaling of the important plant hormone gibberellin, and achieved promising results that reveal interesting insight into the biosynthesis and signaling of gibberellin.
The results show that our method is highly effective in finding functional modules from real microarray data. Our application on Arabidopsis leads to the discovery of the largest number of annotated Arabidopsis functional modules in the literature. Given the high statistical significance of functional enrichment and the agreement between cis-regulatory and functional annotations, we believe our Arabidopsis gene modules can be used to predict the functions of unknown genes in Arabidopsis, and to understand the regulatory mechanisms of many genes.
已经成功构建了几个大规模的基因共表达网络,用于预测拟南芥(Arabidopsis thaliana)中的基因功能模块和顺式调控元件。然而,这些网络通常是专门构建和分析的。在这项研究中,我们提出了一种完全无参数且系统的方法,用于构建基因共表达网络以及预测功能模块和顺式调控元件。
我们的新方法包括一个自动化的网络构建算法、一种无参数的功能模块预测程序以及一种适合于共识扫描的策略,无需先验知识即可找到已知的顺式调控元件,该策略允许基序的简并度在一定范围内变化。我们将该方法应用于研究大量的拟南芥基因表达微阵列数据。我们估计我们的共表达网络的准确率约为 94%,并且具有与其他生物网络相似的拓扑性质,例如具有无标度特性和高聚类系数。值得注意的是,在大小至少为 20 的约 300 个预测模块中,有 88%至少具有一个显著富集的功能,其中包括几个极其显著的功能(核糖体,p < 1E-300,光合膜,p < 1.3E-137,蛋白酶体复合物,p < 5.9E-126)。此外,我们能够预测 66.7%的模块的顺式调控元件,并且富集的顺式调控元件与富集的功能术语之间的关联通常可以通过文献来证实。总的来说,我们的结果比以前在类似数据集上进行的几项研究的结果更为显著。最后,我们利用共表达网络剖析了 19 个参与植物激素赤霉素代谢和信号转导的拟南芥基因的启动子,取得了令人鼓舞的结果,这些结果揭示了赤霉素生物合成和信号转导的有趣见解。
结果表明,我们的方法在从真实微阵列数据中寻找功能模块方面非常有效。我们在拟南芥上的应用导致了文献中发现的最大数量的注释拟南芥功能模块。鉴于功能富集的高度统计学意义以及顺式调控和功能注释之间的一致性,我们相信我们的拟南芥基因模块可以用于预测拟南芥中未知基因的功能,并深入了解许多基因的调控机制。