Molecular Biology Division, National Institute of Malaria Research, Sector-8, Dwarka, New Delhi, India.
Malar J. 2011 Dec 19;10:374. doi: 10.1186/1475-2875-10-374.
Multi-drug resistance and severe/complicated cases are the emerging phenotypes of vivax malaria, which may deteriorate current anti-malarial control measures. The emergence of these phenotypes could be associated with either of the two Plasmodium vivax lineages. The two lineages had been categorized as Old World and New World, based on geographical sub-division and genetic and phenotypical markers. This study revisited the lineage hypothesis of P. vivax by typing the distribution of lineages among global isolates and evaluated their genetic relatedness using a panel of new mini-satellite markers.
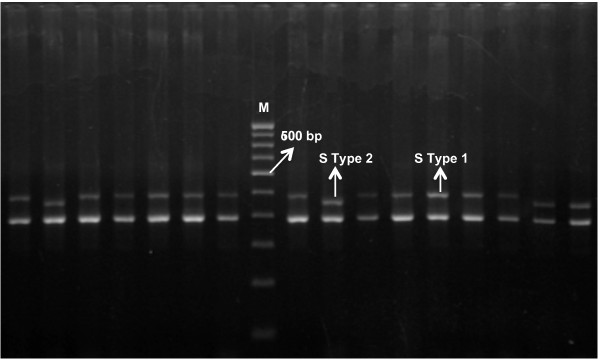
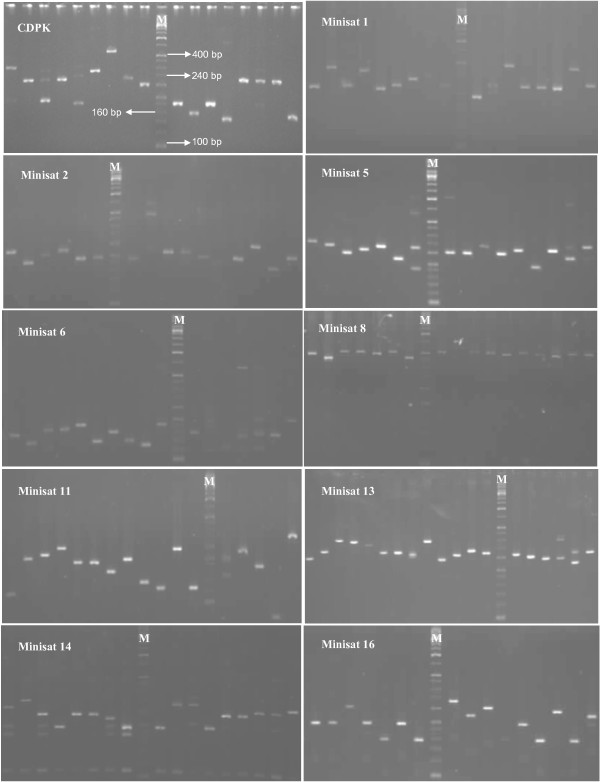
18S SSU rRNA S-type gene was amplified from 420 Plasmodium vivax field isolates collected from different geographical regions of India, Thailand and Colombia as well as four strains each of P. vivax originating from Nicaragua, Panama, Thailand (Pak Chang), and Vietnam (ONG). A mini-satellite marker panel was then developed to understand the population genetic parameters and tested on a sample subset of both lineages.
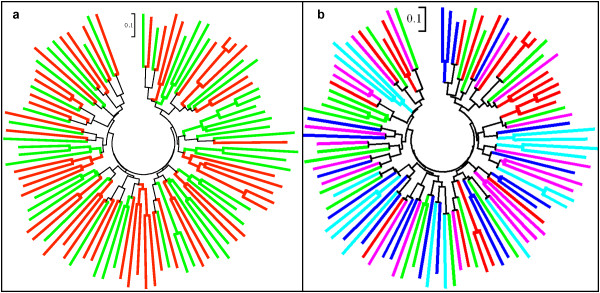
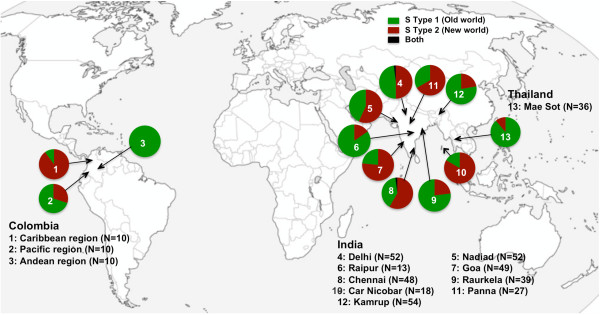
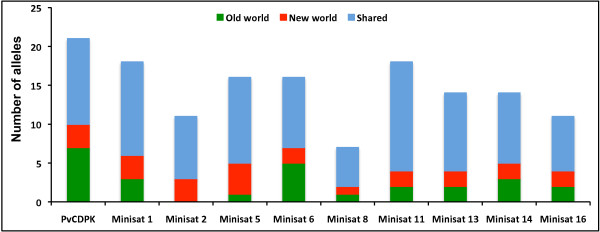
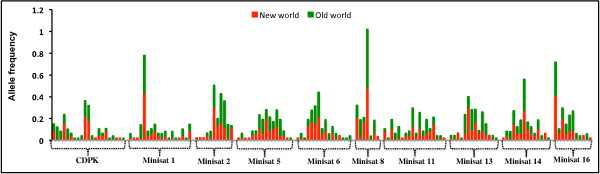
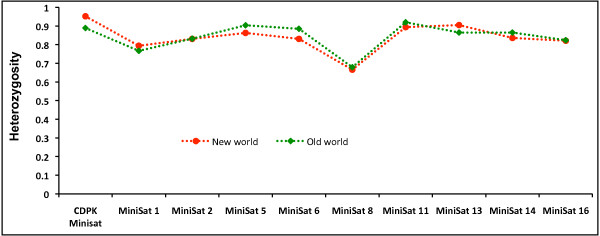
18S SSU rRNA S-type gene typing revealed the distribution of both lineages (Old World and New World) in all geographical regions. However, distribution of Plasmodium vivax lineages was highly variable in every geographical region. The lack of geographical sub-division between lineages suggests that both lineages are globally distributed. Ten mini-satellites were scanned from the P. vivax genome sequence; these tandem repeats were located in eight of the chromosomes. Mini-satellites revealed substantial allelic diversity (7-21, AE = 14.6 ± 2.0) and heterozygosity (He = 0.697-0.924, AE = 0.857 ± 0.033) per locus. Mini-satellite comparison between the two lineages revealed high but similar pattern of genetic diversity, allele frequency, and high degree of allele sharing. A Neighbour-Joining phylogenetic tree derived from genetic distance data obtained from ten mini-satellites also placed both lineages together in every cluster.
The global lineage distribution, lack of genetic distance, similar pattern of genetic diversity, and allele sharing strongly suggested that both lineages are a single species and thus new emerging phenotypes associated with vivax malaria could not be clearly classified as belonging to a particular lineage on basis of their geographical origin.
多重耐药和重症/复杂病例是间日疟的新兴表型,可能会使当前的抗疟控制措施恶化。这些表型的出现可能与间日疟原虫的两个谱系之一有关。这两个谱系根据地理分区以及遗传和表型标记被归类为旧世界和新世界。本研究通过对全球分离株的谱系分布进行分型,重新考察了间日疟原虫的谱系假说,并使用一组新的微卫星标记评估了它们的遗传相关性。
从印度、泰国和哥伦比亚不同地理区域采集的 420 株间日疟原虫现场分离株以及来自尼加拉瓜、巴拿马、泰国(Pak Chang)和越南(ONG)的每株间日疟原虫 4 株中扩增 18S SSU rRNA S 型基因。然后开发了一个微卫星标记面板来了解种群遗传参数,并在两个谱系的样本子集中进行了测试。
18S SSU rRNA S 型基因分型显示,两个谱系(旧世界和新世界)在所有地理区域均有分布。然而,每个地理区域间日疟原虫谱系的分布差异很大。谱系之间没有明显的地理分区表明两个谱系在全球范围内分布。从间日疟原虫基因组序列中扫描了 10 个微卫星,这些串联重复位于 8 条染色体中。微卫星揭示了丰富的等位基因多样性(7-21,AE = 14.6 ± 2.0)和杂合度(He = 0.697-0.924,AE = 0.857 ± 0.033)。两个谱系之间的微卫星比较显示,遗传多样性、等位基因频率和等位基因共享程度都很高,但模式相似。基于 10 个微卫星获得的遗传距离数据构建的邻接法系统发育树也将两个谱系放在了每个聚类中。
全球谱系分布、遗传距离缺失、相似的遗传多样性模式以及等位基因共享强烈表明,两个谱系是一个单一的物种,因此与间日疟相关的新出现的表型不能根据其地理起源明确归类为特定谱系。