Moschopoulos Charalampos N, Pavlopoulos Georgios A, Iacucci Ernesto, Aerts Jan, Likothanassis Spiridon, Schneider Reinhard, Kossida Sophia
Bioinformatics & Medical Informatics Team, Biomedical Research Foundation, Academy of Athens, Soranou Efessiou 4, 11527 Athens, Greece.
BMC Res Notes. 2011 Dec 20;4:549. doi: 10.1186/1756-0500-4-549.
Protein-Protein interactions (PPI) play a key role in determining the outcome of most cellular processes. The correct identification and characterization of protein interactions and the networks, which they comprise, is critical for understanding the molecular mechanisms within the cell. Large-scale techniques such as pull down assays and tandem affinity purification are used in order to detect protein interactions in an organism. Today, relatively new high-throughput methods like yeast two hybrid, mass spectrometry, microarrays, and phage display are also used to reveal protein interaction networks.
In this paper we evaluated four different clustering algorithms using six different interaction datasets. We parameterized the MCL, Spectral, RNSC and Affinity Propagation algorithms and applied them to six PPI datasets produced experimentally by Yeast 2 Hybrid (Y2H) and Tandem Affinity Purification (TAP) methods. The predicted clusters, so called protein complexes, were then compared and benchmarked with already known complexes stored in published databases.
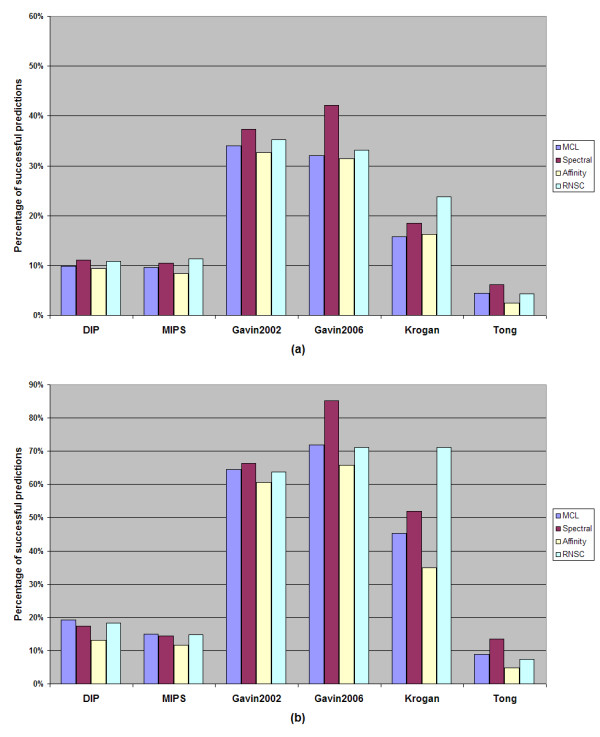
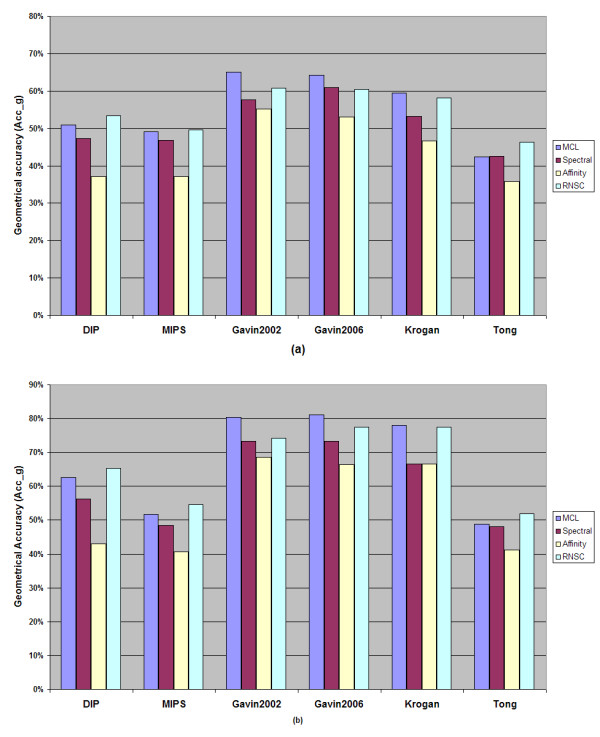
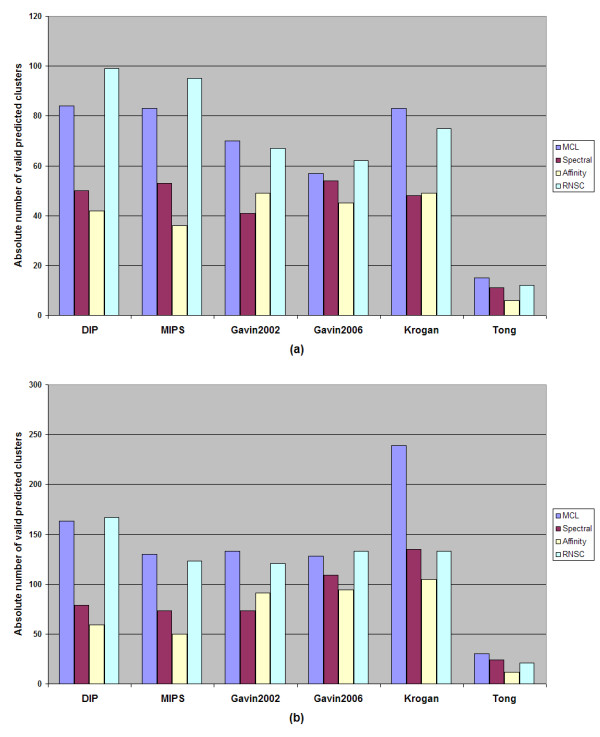
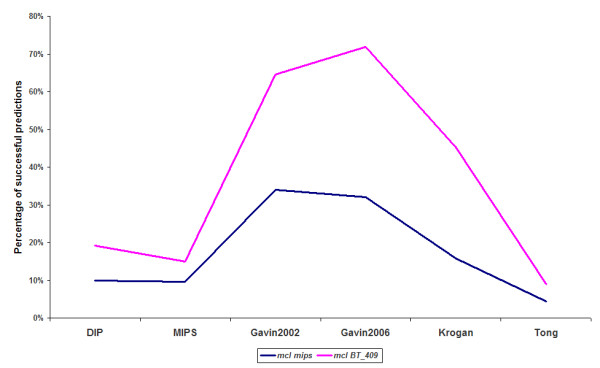
While results may differ upon parameterization, the MCL and RNSC algorithms seem to be more promising and more accurate at predicting PPI complexes. Moreover, they predict more complexes than other reviewed algorithms in absolute numbers. On the other hand the spectral clustering algorithm achieves the highest valid prediction rate in our experiments. However, it is nearly always outperformed by both RNSC and MCL in terms of the geometrical accuracy while it generates the fewest valid clusters than any other reviewed algorithm. This article demonstrates various metrics to evaluate the accuracy of such predictions as they are presented in the text below. Supplementary material can be found at: http://www.bioacademy.gr/bioinformatics/projects/ppireview.htm.
蛋白质-蛋白质相互作用(PPI)在决定大多数细胞过程的结果中起着关键作用。正确识别和表征蛋白质相互作用及其所构成的网络,对于理解细胞内的分子机制至关重要。为了检测生物体中的蛋白质相互作用,人们使用了诸如下拉分析和串联亲和纯化等大规模技术。如今,相对较新的高通量方法,如酵母双杂交、质谱、微阵列和噬菌体展示,也被用于揭示蛋白质相互作用网络。
在本文中,我们使用六个不同的相互作用数据集评估了四种不同的聚类算法。我们对马尔可夫聚类算法(MCL)、谱聚类算法、基于随机邻居搜索的聚类算法(RNSC)和亲和传播算法进行了参数化,并将它们应用于通过酵母双杂交(Y2H)和串联亲和纯化(TAP)方法实验产生的六个PPI数据集。然后,将预测的聚类,即所谓的蛋白质复合物,与已发表数据库中存储的已知复合物进行比较和基准测试。
虽然参数化后结果可能不同,但MCL和RNSC算法在预测PPI复合物方面似乎更有前景且更准确。此外,它们预测的复合物绝对数量比其他所审查的算法更多。另一方面,谱聚类算法在我们的实验中实现了最高的有效预测率。然而,在几何准确性方面,它几乎总是不如RNSC和MCL,同时它产生的有效聚类比任何其他所审查的算法都少。本文展示了各种指标来评估此类预测的准确性,如下文所述。补充材料可在以下网址找到:http://www.bioacademy.gr/bioinformatics/projects/ppireview.htm。