Vincent Ajoy, Wright Tom, Day Megan A, Westall Carol A, Héon Elise
Department of Ophthalmology and Vision Sciences, The Hospital for Sick Children, University of Toronto, 555 University Avenue, Toronto, Ontario, Canada.
Mol Vis. 2011;17:3262-70. Epub 2011 Dec 15.
To report, for the first time, that X-linked incomplete congenital stationary night blindness (CSNB2A) and Åland island eye disease (AIED) phenotypes coexist in a molecularly confirmed pedigree and to present novel phenotypic characteristics of calcium channel alpha-1F subunit gene (CACNA1F)-related disease.
Two affected subjects (the proband and his maternal grandfather) and an unaffected obligate carrier (the proband's mother) underwent detailed ophthalmological evaluation, fundus autofluorescence imaging, and spectral-domain optical coherence tomography. Goldmann visual field assessment and full-field electroretinogram (ERG) were performed in the two affected subjects, and multichannel flash visual evoked potential was performed on the proband. Scotopic 15 Hz flicker ERG series were performed in both affected subjects to evaluate the function of the slow and fast rod pathways. Haplotype analysis using polymorphic microsatellite markers flanking CACNA1F was performed in all three family members. The proband's DNA was sequenced for mutations in the coding sequence of CACNA1F and nyctalopin (NYX) genes. Segregation analysis was performed in the family.
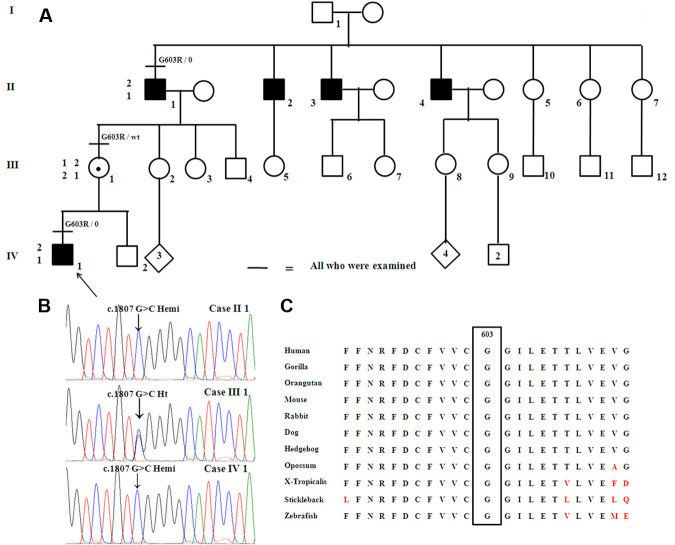
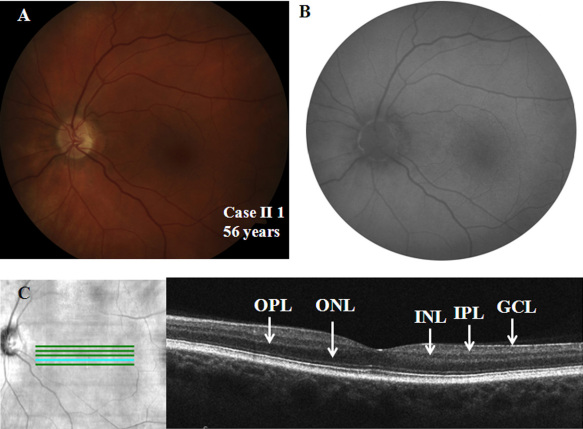
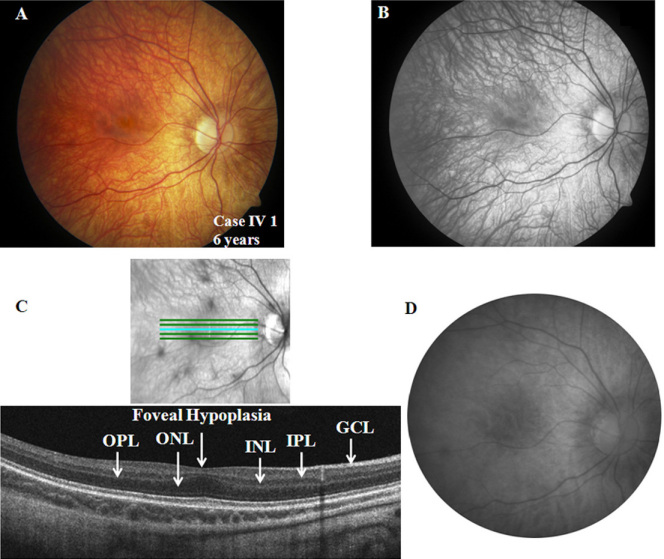
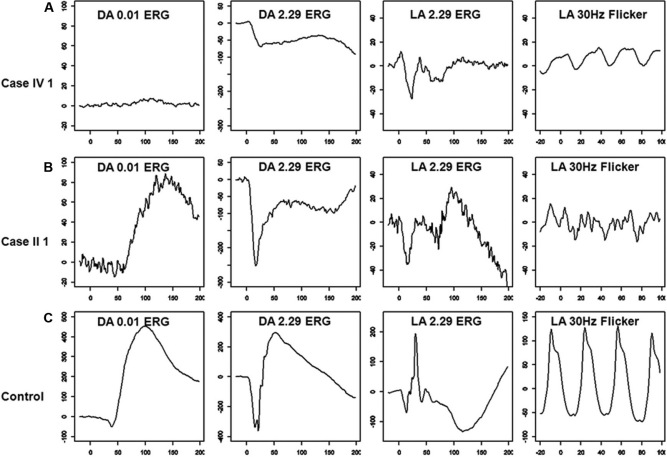
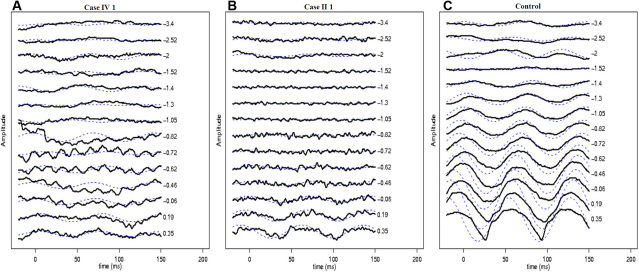
Both affected subjects had symptoms of nonprogressive nyctalopia since childhood, while the proband also had photophobia. Both cases had a distance visual acuity of 20/50 or better in each eye, normal contrast sensitivity, and an incomplete type of Schubert-Bornschein ERGs. The proband also had high myopia, a mild red-green color deficit, hypopigmented fundus, and foveal hypoplasia with no evidence of chiasmal misrouting. Spectral-domain optical coherence tomography confirmed the presence of foveal hypoplasia in the proband. The clinical phenotype of the proband and his maternal grandfather fit the clinical description of AIED and CSNB2A, respectively. The fundus autofluorescence and the visual fields were normal in both cases; the scotopic 15 Hz flicker ERG demonstrated only fast rod pathway activity in both. Both affected cases shared the same haplotype across CACNA1F. The proband carried a novel hemizygous c.1807G>C mutation (p.G603R) in the CACNA1F gene. The change segregated with the disease phenotypes and was not identified in 360 control chromosomes. No mutations were identified in NYX.
This report of a missense mutation in CACNA1F causing AIED and CSNB2A phenotypes in a family confirms that both diseases are allelic and that other genetic or environmental modifiers influence the expression of CACNA1F. This is the first report to suggest that in CACNA1F-related disease, the rod system activity is predominantly from the fast rod pathways.
首次报告在一个经分子确认的家系中,X连锁不完全先天性静止性夜盲(CSNB2A)和奥兰岛眼病(AIED)表型共存,并呈现钙通道α-1F亚基基因(CACNA1F)相关疾病的新表型特征。
两名受影响的受试者(先证者及其外祖父)和一名未受影响的 obligate 携带者(先证者的母亲)接受了详细的眼科评估、眼底自发荧光成像和光谱域光学相干断层扫描。对两名受影响的受试者进行了 Goldmann 视野评估和全视野视网膜电图(ERG)检查,对先证者进行了多通道闪光视觉诱发电位检查。在两名受影响的受试者中进行了暗视15Hz闪烁ERG系列检查,以评估慢和快视杆通路的功能。使用位于CACNA1F侧翼的多态微卫星标记对所有三名家庭成员进行单倍型分析。对先证者的DNA进行测序,以检测CACNA1F和夜盲蛋白(NYX)基因编码序列中的突变。在该家族中进行了分离分析。
两名受影响的受试者自童年起均有非进行性夜盲症状,而先证者还畏光。两例患者每只眼的远视力均为20/50或更好,对比敏感度正常,ERG为Schubert-Bornschein不完全型。先证者还患有高度近视、轻度红绿色觉异常、眼底色素减退和黄斑发育不全,无视交叉错路的证据。光谱域光学相干断层扫描证实先证者存在黄斑发育不全。先证者及其外祖父的临床表型分别符合AIED和CSNB2A的临床描述。两例患者的眼底自发荧光和视野均正常;暗视15Hz闪烁ERG显示两者均只有快视杆通路活动。两名受影响的病例在CACNA1F上共享相同的单倍型。先证者在CACNA1F基因中携带一个新的半合子c.1807G>C突变(p.G603R)。该变化与疾病表型分离,在360条对照染色体中未发现。在NYX中未发现突变。
本报告中一个家系中CACNA1F的错义突变导致AIED和CSNB2A表型,证实这两种疾病是等位基因,并且其他遗传或环境修饰因子影响CACNA1F的表达。这是首次表明在CACNA1F相关疾病中,视杆系统活动主要来自快视杆通路的报告。