School of Informatics, Indiana University Purdue University, Indianapolis, IN, USA.
J Biomol Struct Dyn. 2012;29(4):799-813. doi: 10.1080/073911012010525022.
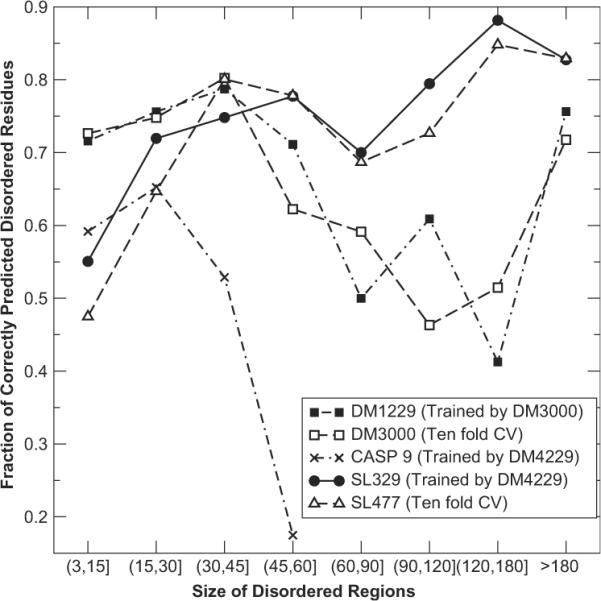
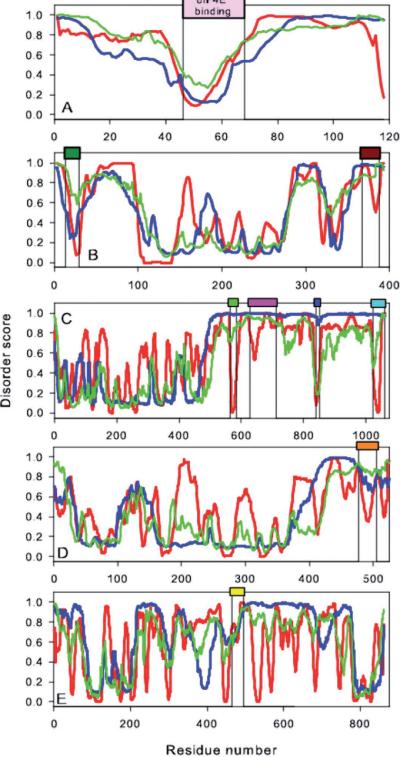
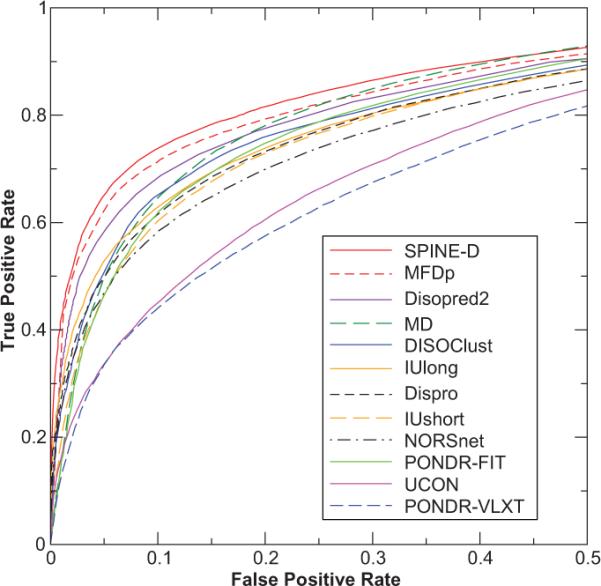
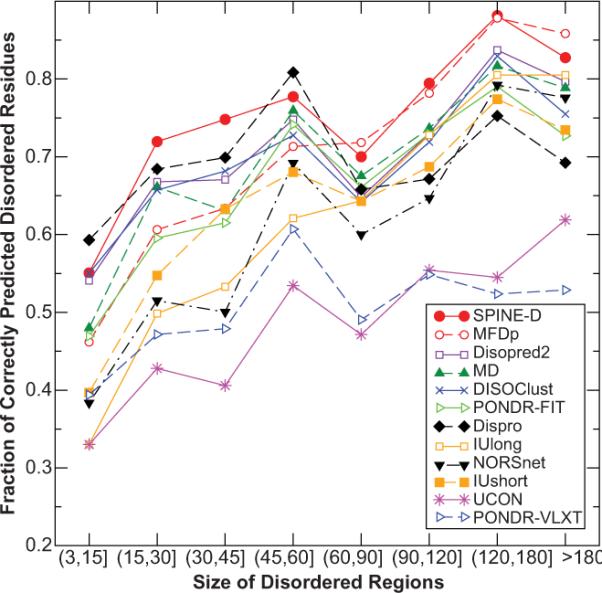
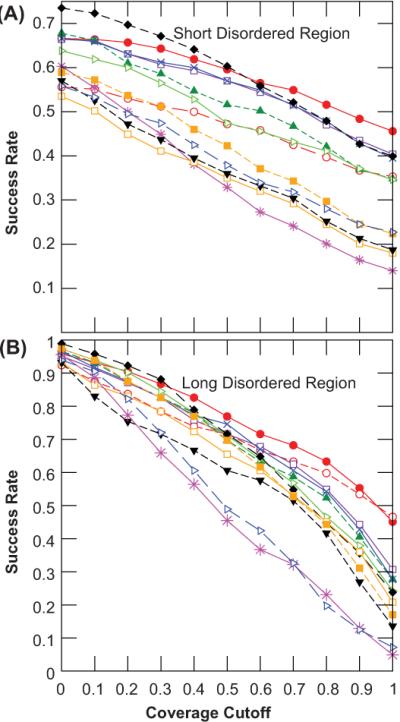
Short and long disordered regions of proteins have different preference for different amino acid residues. Different methods often have to be trained to predict them separately. In this study, we developed a single neural-network-based technique called SPINE-D that makes a three-state prediction first (ordered residues and disordered residues in short and long disordered regions) and reduces it into a two-state prediction afterwards. SPINE-D was tested on various sets composed of different combinations of Disprot annotated proteins and proteins directly from the PDB annotated for disorder by missing coordinates in X-ray determined structures. While disorder annotations are different according to Disprot and X-ray approaches, SPINE-D's prediction accuracy and ability to predict disorder are relatively independent of how the method was trained and what type of annotation was employed but strongly depend on the balance in the relative populations of ordered and disordered residues in short and long disordered regions in the test set. With greater than 85% overall specificity for detecting residues in both short and long disordered regions, the residues in long disordered regions are easier to predict at 81% sensitivity in a balanced test dataset with 56.5% ordered residues but more challenging (at 65% sensitivity) in a test dataset with 90% ordered residues. Compared to eleven other methods, SPINE-D yields the highest area under the curve (AUC), the highest Mathews correlation coefficient for residue-based prediction, and the lowest mean square error in predicting disorder contents of proteins for an independent test set with 329 proteins. In particular, SPINE-D is comparable to a meta predictor in predicting disordered residues in long disordered regions and superior in short disordered regions. SPINE-D participated in CASP 9 blind prediction and is one of the top servers according to the official ranking. In addition, SPINE-D was examined for prediction of functional molecular recognition motifs in several case studies.
蛋白质的短和长无序区域对不同的氨基酸残基有不同的偏好。通常需要针对它们分别训练不同的方法。在这项研究中,我们开发了一种基于单一神经网络的技术,称为 SPINE-D,它首先进行三状态预测(有序残基和短、长无序区域中的无序残基),然后将其简化为二状态预测。SPINE-D 在各种由不同 Disprot 注释蛋白质和直接从 PDB 中注释的蛋白质组成的集合上进行了测试,这些蛋白质的无序状态是通过 X 射线确定结构中缺失坐标来注释的。虽然 Disprot 和 X 射线方法的无序注释不同,但 SPINE-D 的预测准确性和无序预测能力相对独立于训练方法和使用的注释类型,但强烈依赖于测试集中短和长无序区域中有序和无序残基的相对比例的平衡。对于检测短和长无序区域中的残基,总体特异性大于 85%,在具有 56.5%有序残基的平衡测试数据集(81%的灵敏度)中,长无序区域中的残基更容易预测,但在具有 90%有序残基的测试数据集(65%的灵敏度)中更具挑战性。与其他 11 种方法相比,SPINE-D 在独立测试集上具有最高的曲线下面积(AUC)、基于残基预测的最高 Matthews 相关系数和最低的均方误差,用于预测蛋白质的无序含量。特别是,SPINE-D 在预测长无序区域中的无序残基方面与元预测器相当,在短无序区域中表现更好。SPINE-D 参加了 CASP 9 盲测,根据官方排名,它是排名最高的服务器之一。此外,SPINE-D 还在几个案例研究中用于预测功能分子识别基序。