Division of Immunohematology and Transfusion Medicine, Dept. Oncology-Hematology, Ospedali Riuniti, Bergamo, Italy.
Mediterr J Hematol Infect Dis. 2011;3(1):e2011068. doi: 10.4084/MJHID.2011.068. Epub 2011 Dec 21.
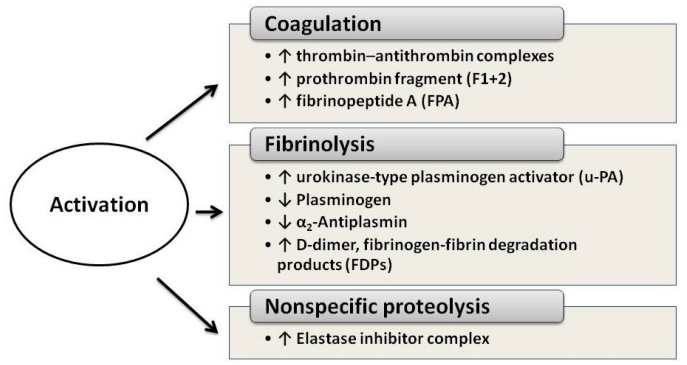
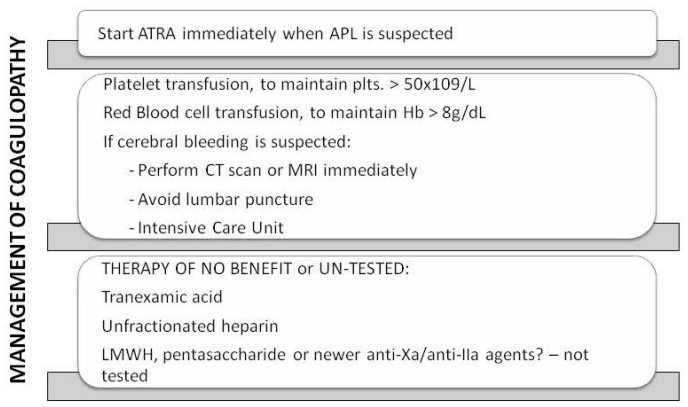
Acute promyelocytic leukemia (APL) is a distinct subtype of myeloid leukemia characterized by t(15;17) chromosomal translocation, which involves the retinoic acid receptor-alpha (RAR-alpha). APL typically presents with a life-threatening hemorrhagic diathesis. Before the introduction of all-trans retinoic acid (ATRA) for the cure of APL, fatal hemorrhages due, at least in part, to the APL-associated coagulopathy, were a major cause of induction remission failure. The laboratory abnormalities of blood coagulation found in these patients indicate the occurrence of a hypercoagulable state. Major determinants of the coagulopathy of APL are endogenous factors expressed by the leukemic cells, including procoagulant factors, fibrinolytic proteins, and non-specific proteolytic enzymes. In addition, these cells have an increased capacity to adhere to the vascular endothelium, and to secrete inflammatory cytokines [i.e. interleukin-1beta (IL-1beta) and tumor necrosis factor (TNF-alpha)], which in turn stimulate the expression of prothrombotic activities by endothelial cells and leukocytes. ATRA can interfere with each of the principal hemostatic properties of the leukemic cell, thus reducing the APL cell procoagulant potential, in parallel to the induction of cellular differentiation. This effect occurs in vivo, in the bone marrow of APL patients receiving ATRA, and is associated with the improvement of the bleeding symptoms. Therapy with arsenic trioxide (ATO) also beneficially affects coagulation in APL. However, early deaths from bleeding still remain a major problem in APL and further research is required in this field. In this review, we will summarize our current knowledge of the pathogenesis of the APL-associated coagulopathy and will overview the therapeutic approaches for the management of this complication.
急性早幼粒细胞白血病(APL)是一种独特的髓系白血病亚型,其特征为 t(15;17)染色体易位,涉及维甲酸受体-α(RAR-α)。APL 通常表现为危及生命的出血倾向。在全反式维甲酸(ATRA)用于治疗 APL 之前,由于 APL 相关的凝血障碍,至少部分原因导致致命性出血,是诱导缓解失败的主要原因。这些患者血液凝血的实验室异常表明发生了高凝状态。APL 凝血障碍的主要决定因素是白血病细胞表达的内源性因子,包括促凝因子、纤维蛋白溶解蛋白和非特异性蛋白酶。此外,这些细胞具有增加的黏附血管内皮和分泌炎症细胞因子的能力[即白细胞介素-1β(IL-1β)和肿瘤坏死因子(TNF-α)],这反过来又刺激内皮细胞和白细胞表达促血栓形成活性。ATRA 可以干扰白血病细胞的每一个主要止血特性,从而降低 APL 细胞的促凝潜能,同时诱导细胞分化。这种作用发生在体内,在接受 ATRA 的 APL 患者的骨髓中,与出血症状的改善相关。三氧化二砷(ATO)治疗也对 APL 中的凝血产生有益影响。然而,APL 患者仍有早期因出血而死亡的主要问题,因此需要在这一领域开展进一步的研究。在这篇综述中,我们将总结我们目前对 APL 相关凝血障碍发病机制的认识,并概述管理这种并发症的治疗方法。