Airways Disease Section, National Heart and Lung Institute, Imperial College London, London, UK.
J Inflamm (Lond). 2012 Jan 12;9(1):1. doi: 10.1186/1476-9255-9-1.
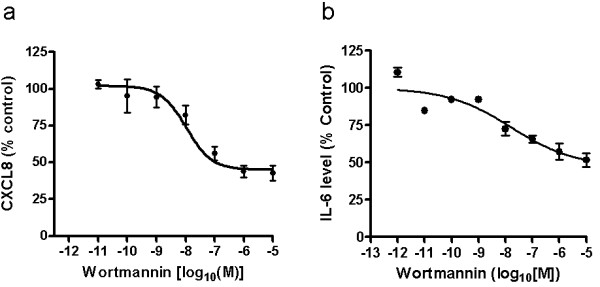
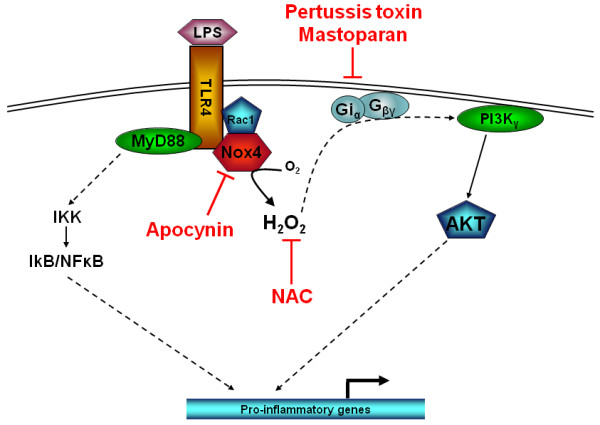
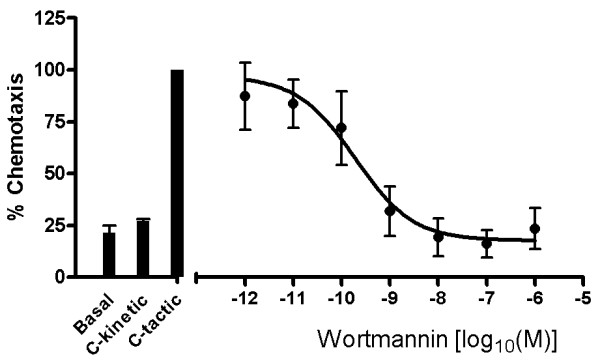
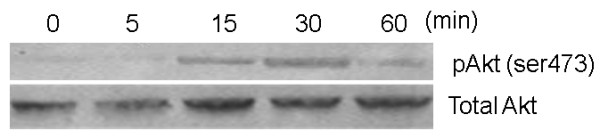
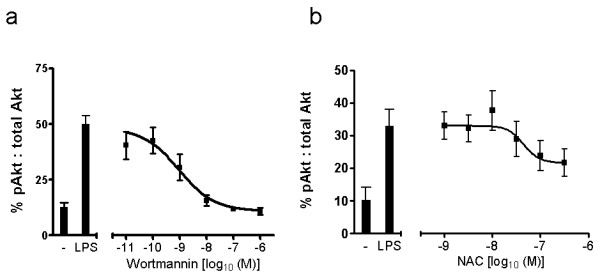
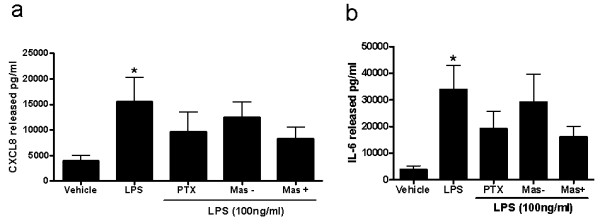
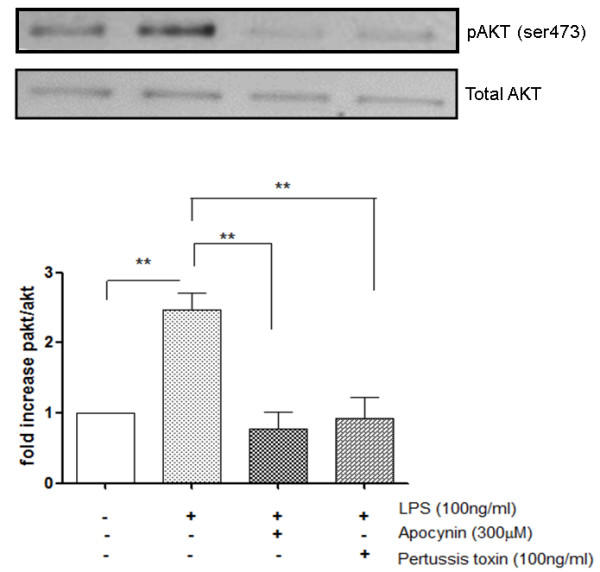
COPD is a disease of innate immunity and bacterial infections are a dominant cause of exacerbations in the later stages resulting in poor health and high mortality. The pathogen-associated molecular pattern (PAMP) lipopolysaccharide (LPS) is sensed by immune cells through activation of the toll-like receptor 4 (TLR4). This leads to the activation of NADPH oxidase (NOX) and NF-κB which together drive COPD inflammation. In this study we show in human PBMCs that LPS stimulated proinflammatory cytokine release (CXCL8 and IL6) was inhibited by approximately 50% by the broad specificity phosphatidylinositol 3-kinase (PI3K) inhibitor, wortmannin. Our results also demonstrate that activation of PI3K following LPS stimulation is mediated by a NOX4 dependent mechanism releasing endogenous H2O2, as the NOX4 inhibitor apocynin blocked LPS induced AKT phosphorylation. Moreover, LPS-induced PI3K activation was inhibited by the anti-oxidant N-acetylcysteine in a concentration dependent manner (IC50 ~100 μM). In addition, our data demonstrated that inhibition of small G proteins, by pre-treatment with pertussis toxin, inhibited LPS-induced AKT phosphorylation. Furthermore, the G-protein inhibitors pertussis toxin and mastoparan both inhibited LPS-induced CXCL8 and IL-6 release by approximately 50%. Together, these data indicate there is a mechanism in human PBMCs where TLR4 activation by LPS leads to ROS generation through NOX4 and activation of the PI3K pathway. This effect is apparently mediated through small G proteins facilitating the release of pro-inflammatory cytokines.
COPD 是一种先天免疫疾病,细菌感染是后期加重的主要原因,导致健康状况不佳和高死亡率。病原体相关分子模式 (PAMP) 脂多糖 (LPS) 通过免疫细胞中 Toll 样受体 4 (TLR4) 的激活来感知。这导致 NADPH 氧化酶 (NOX) 和 NF-κB 的激活,两者共同驱动 COPD 炎症。在这项研究中,我们在人类 PBMC 中表明,LPS 刺激的促炎细胞因子释放 (CXCL8 和 IL6) 被广谱特异性磷脂酰肌醇 3-激酶 (PI3K) 抑制剂wortmannin 抑制约 50%。我们的结果还表明,LPS 刺激后 PI3K 的激活是通过 NOX4 依赖性机制介导的,该机制释放内源性 H2O2,因为 NOX4 抑制剂 apocynin 阻断了 LPS 诱导的 AKT 磷酸化。此外,LPS 诱导的 PI3K 激活被抗氧化剂 N-乙酰半胱氨酸以浓度依赖的方式抑制 (IC50~100 μM)。此外,我们的数据表明,通过预先用百日咳毒素处理来抑制小 G 蛋白,可抑制 LPS 诱导的 AKT 磷酸化。此外,G 蛋白抑制剂百日咳毒素和 mastoparan 均可抑制 LPS 诱导的 CXCL8 和 IL-6 释放约 50%。综上所述,这些数据表明,在人类 PBMC 中存在一种机制,即 LPS 通过 TLR4 激活导致通过 NOX4 和激活 PI3K 途径产生 ROS。这种作用显然是通过小 G 蛋白介导的,促进了促炎细胞因子的释放。