Bowles Center for Alcohol Studies, School of Medicine, Chapel Hill, NC 27599, USA.
J Neuroinflammation. 2012 Jan 12;9:5. doi: 10.1186/1742-2094-9-5.
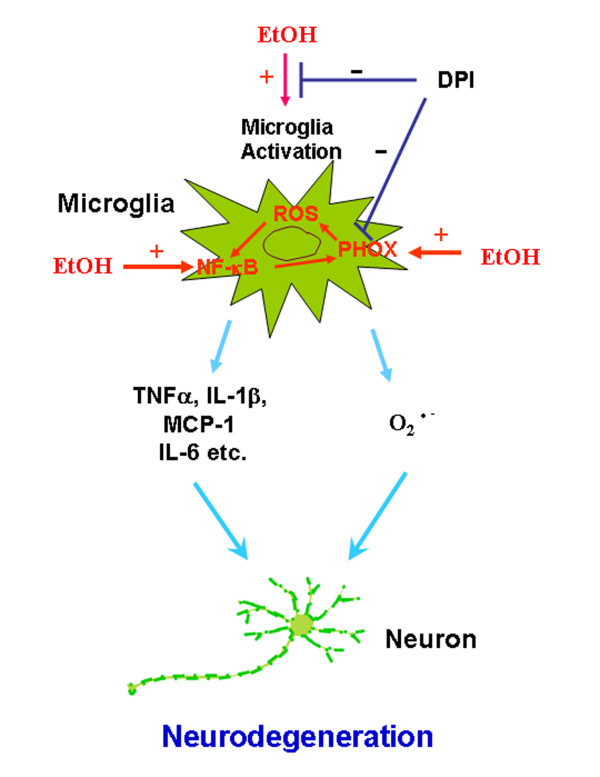
Activation of microglia causes the production of proinflammatory factors and upregulation of NADPH oxidase (NOX) that form reactive oxygen species (ROS) that lead to neurodegeneration. Previously, we reported that 10 daily doses of ethanol treatment induced innate immune genes in brain. In the present study, we investigate the effects of chronic ethanol on activation of NOX and release of ROS, and their contribution to ethanol neurotoxicity.
Male C57BL/6 and NF-κB enhanced GFP mice were treated intragastrically with water or ethanol (5 g/kg, i.g., 25% ethanol w/v) daily for 10 days. The effects of chronic ethanol on cell death markers (activated caspase-3 and Fluoro-Jade B), microglial morphology, NOX, ROS and NF-κB were examined using real-time PCR, immunohistochemistry and hydroethidine histochemistry. Also, Fluoro-Jade B staining and NOX gp91phox immunohistochemistry were performed in the orbitofrontal cortex (OFC) of human postmortem alcoholic brain and human moderate drinking control brain.
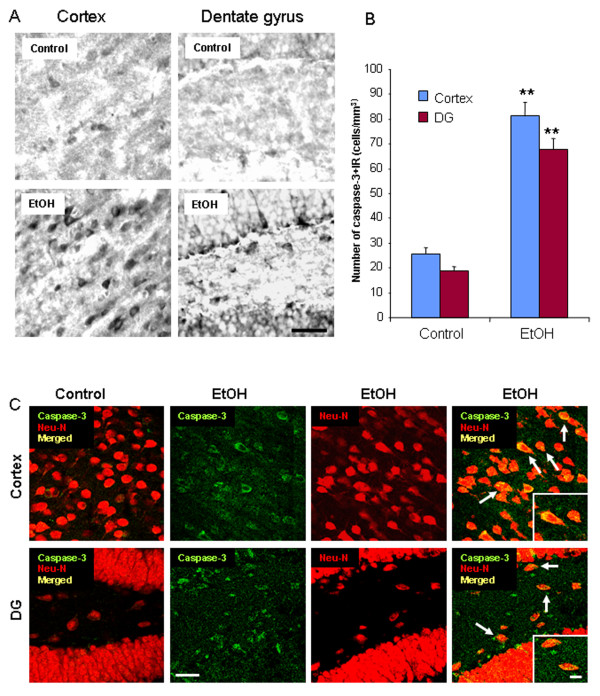
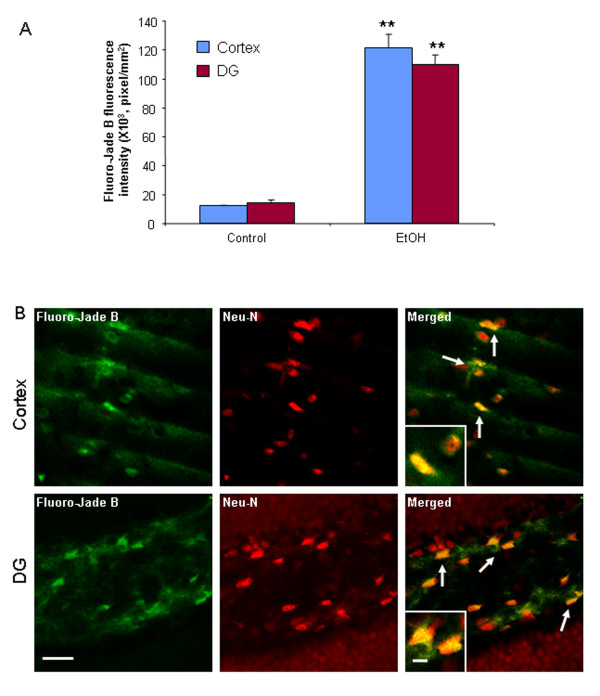
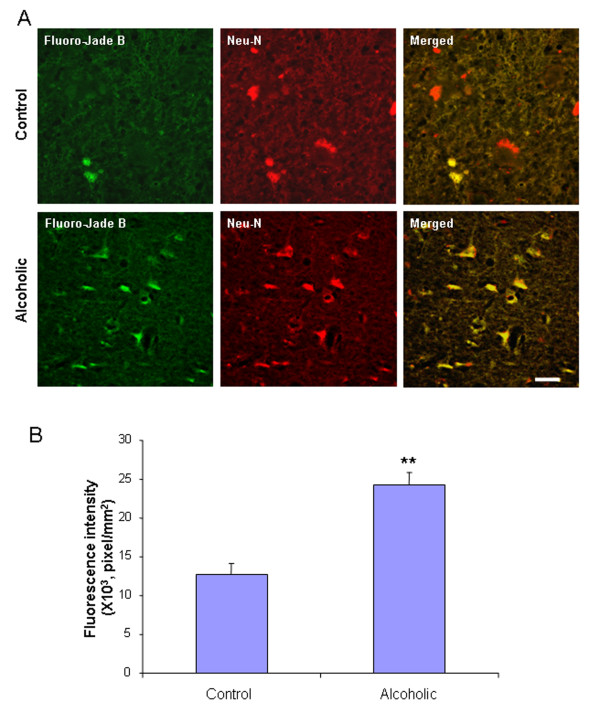
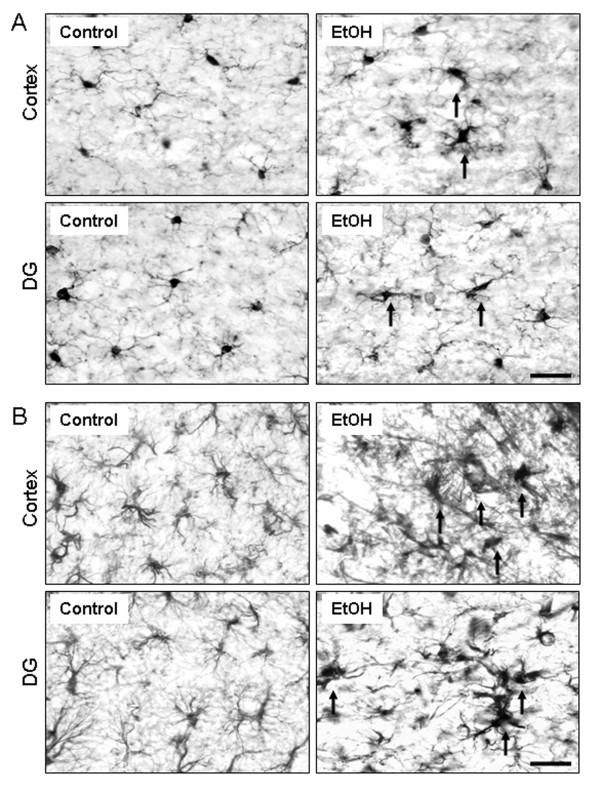
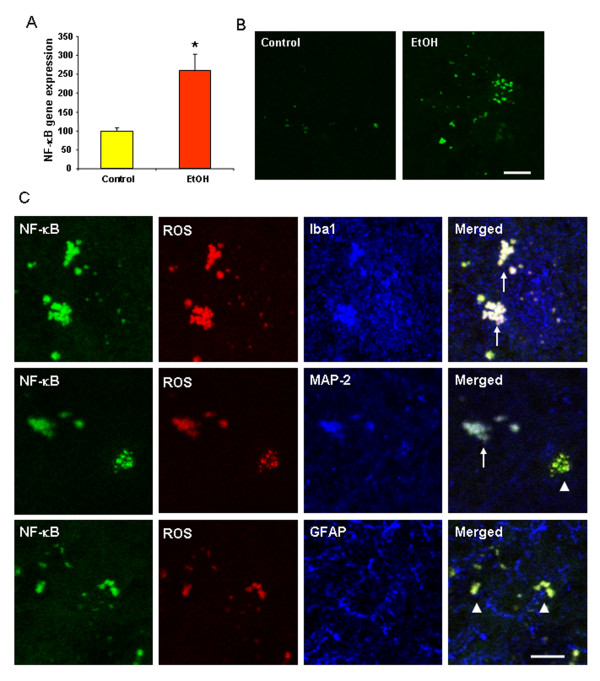
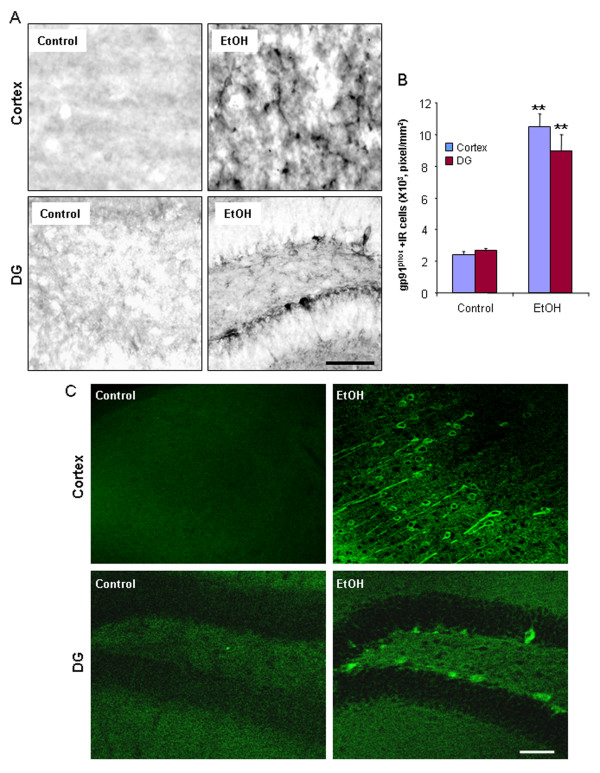
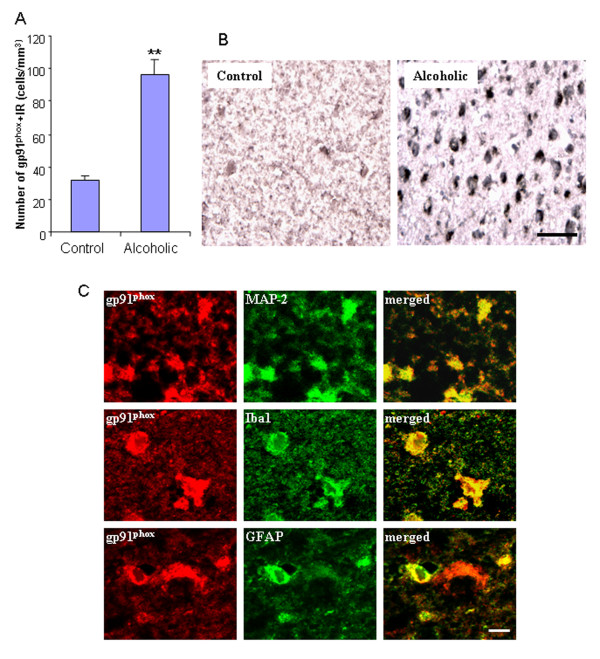
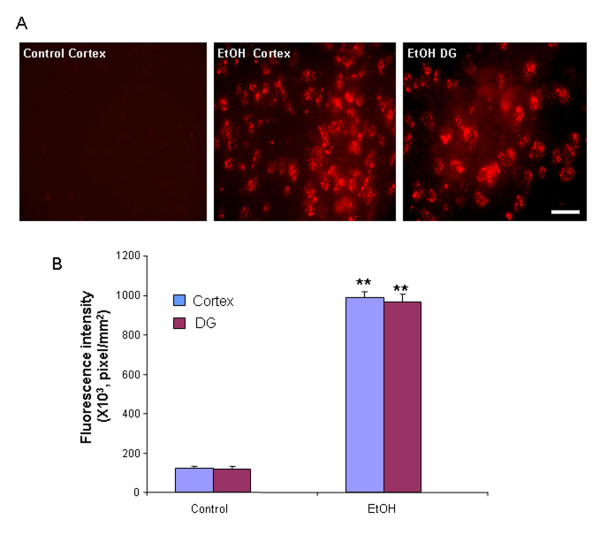
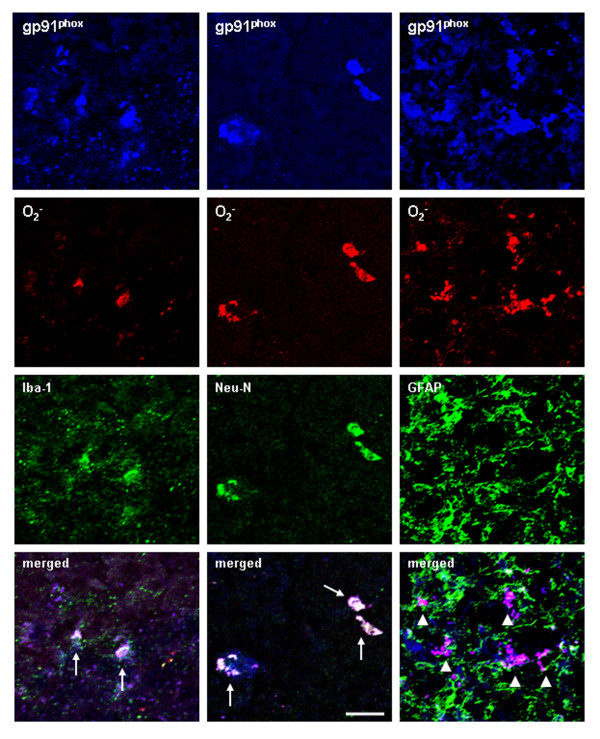
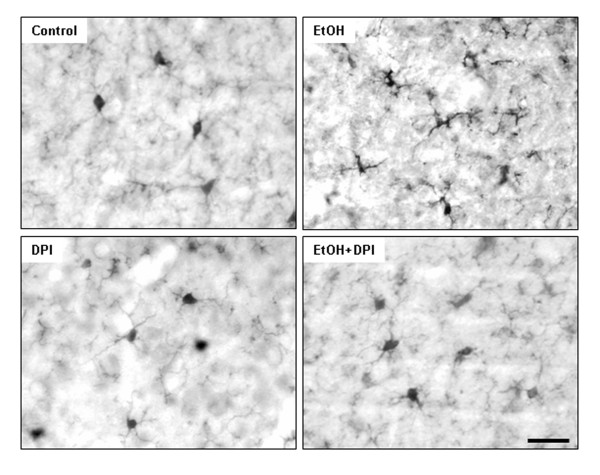
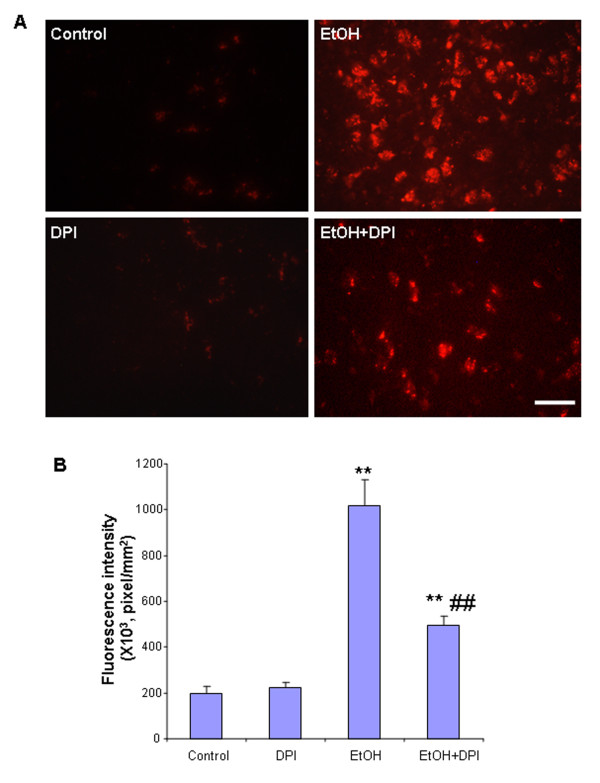
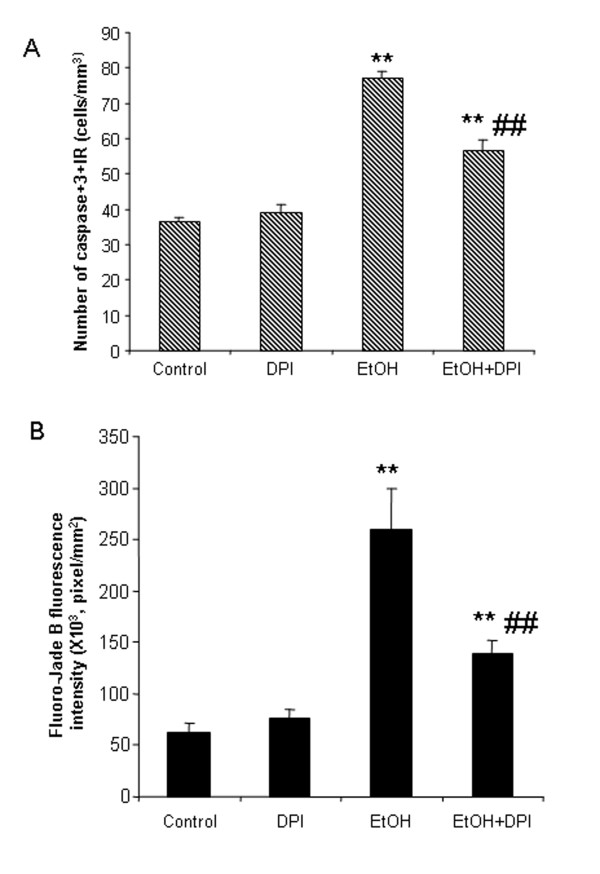
Ethanol treatment of C57BL/6 mice showed increased markers of neuronal death: activated caspase-3 and Fluoro-Jade B positive staining with Neu-N (a neuronal marker) labeling in cortex and dentate gyrus. The OFC of human post-mortem alcoholic brain also showed significantly more Fluoro-Jade B positive cells colocalized with Neu-N, a neuronal marker, compared to the OFC of human moderate drinking control brain, suggesting increased neuronal death in the OFC of human alcoholic brain. Iba1 and GFAP immunohistochemistry showed activated morphology of microglia and astrocytes in ethanol-treated mouse brain. Ethanol treatment increased NF-κB transcription and increased NOX gp91phox at 24 hr after the last ethanol treatment that remained elevated at 1 week. The OFC of human postmortem alcoholic brain also had significant increases in the number of gp91phox + immunoreactive (IR) cells that are colocalized with neuronal, microglial and astrocyte markers. In mouse brain ethanol increased gp91phox expression coincided with increased production of O2- and O2- - derived oxidants. Diphenyleneiodonium (DPI), a NOX inhibitor, reduced markers of neurodegeneration, ROS and microglial activation.
Ethanol activation of microglia and astrocytes, induction of NOX and production of ROS contribute to chronic ethanol-induced neurotoxicity. NOX-ROS and NF-κB signaling pathways play important roles in chronic ethanol-induced neuroinflammation and neurodegeneration.
小胶质细胞的激活会导致促炎因子的产生和 NADPH 氧化酶(NOX)的上调,从而形成活性氧(ROS),导致神经退行性变。此前,我们报道了 10 次每日乙醇处理会诱导大脑中的固有免疫基因。在本研究中,我们研究了慢性乙醇对 NOX 激活和 ROS 释放的影响,以及它们对乙醇神经毒性的贡献。
雄性 C57BL/6 和 NF-κB 增强 GFP 小鼠通过胃内给予水或乙醇(5g/kg,即 25%乙醇 w/v),每天 1 次,连续 10 天。使用实时 PCR、免疫组织化学和羟乙基噻吩组织化学检测慢性乙醇对细胞死亡标志物(活化的 caspase-3 和 Fluoro-Jade B)、小胶质细胞形态、NOX、ROS 和 NF-κB 的影响。此外,还在酒精性脑死后的额眶皮质(OFC)和人类适度饮酒对照脑的额眶皮质中进行了 Fluoro-Jade B 染色和 NOX gp91phox 免疫组织化学染色。
乙醇处理 C57BL/6 小鼠显示神经元死亡标志物增加:活化的 caspase-3 和 Fluoro-Jade B 阳性染色,皮质和齿状回神经元标记物 Neu-N 阳性。与人类适度饮酒对照脑的额眶皮质相比,酒精性脑死后额眶皮质的 Fluoro-Jade B 阳性细胞明显更多,与神经元标记物 Neu-N 共定位,提示人类酒精性脑额眶皮质神经元死亡增加。Iba1 和 GFAP 免疫组织化学显示,乙醇处理后小鼠大脑中小胶质细胞和星形胶质细胞的形态被激活。乙醇处理后 24 小时 NF-κB 转录增加,NOX gp91phox 增加,1 周后仍升高。酒精性脑死后额眶皮质的 gp91phox+免疫反应(IR)细胞数量也显著增加,与神经元、小胶质细胞和星形胶质细胞标记物共定位。在小鼠脑内,乙醇增加 gp91phox 的表达与 O2-和 O2--衍生的氧化剂的产生一致。DPI(一种 NOX 抑制剂)可减少神经退行性变标志物、ROS 和小胶质细胞激活。
乙醇激活小胶质细胞和星形胶质细胞,诱导 NOX 产生 ROS,导致慢性乙醇诱导的神经毒性。NOX-ROS 和 NF-κB 信号通路在慢性乙醇诱导的神经炎症和神经退行性变中起重要作用。