Office of Regulatory Science, Center for Food Safety & Applied Nutrition, U,S, Food & Drug Administration, 5100 Paint Branch Parkway, College Park, MD 20740, USA.
BMC Genomics. 2012 Jan 19;13:32. doi: 10.1186/1471-2164-13-32.
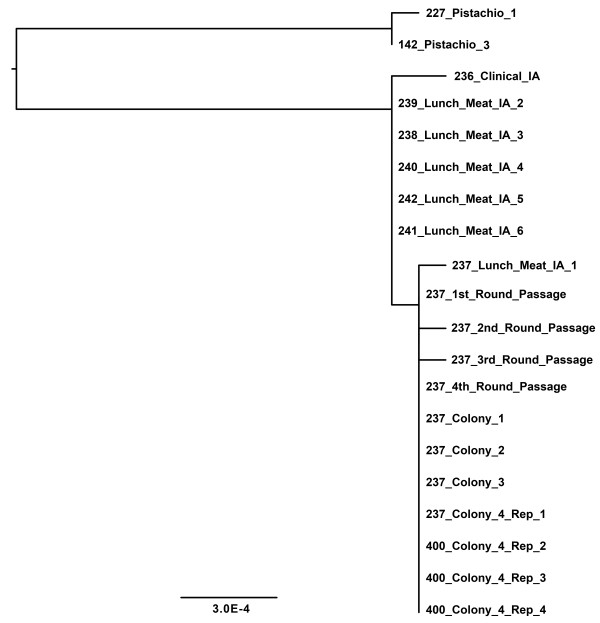
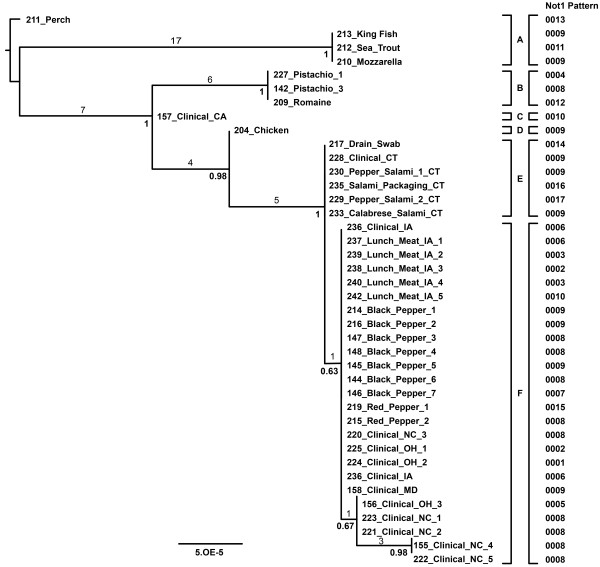
Next-Generation Sequencing (NGS) is increasingly being used as a molecular epidemiologic tool for discerning ancestry and traceback of the most complicated, difficult to resolve bacterial pathogens. Making a linkage between possible food sources and clinical isolates requires distinguishing the suspected pathogen from an environmental background and placing the variation observed into the wider context of variation occurring within a serovar and among other closely related foodborne pathogens. Equally important is the need to validate these high resolution molecular tools for use in molecular epidemiologic traceback. Such efforts include the examination of strain cluster stability as well as the cumulative genetic effects of sub-culturing on these clusters. Numerous isolates of S. Montevideo were shot-gun sequenced including diverse lineage representatives as well as numerous replicate clones to determine how much variability is due to bias, sequencing error, and or the culturing of isolates. All new draft genomes were compared to 34 S. Montevideo isolates previously published during an NGS-based molecular epidemiological case study.
Intraserovar lineages of S. Montevideo differ by thousands of SNPs, that are only slightly less than the number of SNPs observed between S. Montevideo and other distinct serovars. Much less variability was discovered within an individual S. Montevideo clade implicated in a recent foodborne outbreak as well as among individual NGS replicates. These findings were similar to previous reports documenting homopolymeric and deletion error rates with the Roche 454 GS Titanium technology. In no case, however, did variability associated with sequencing methods or sample preparations create inconsistencies with our current phylogenetic results or the subsequent molecular epidemiological evidence gleaned from these data.
Implementation of a validated pipeline for NGS data acquisition and analysis provides highly reproducible results that are stable and predictable for molecular epidemiological applications. When draft genomes are collected at 15×-20× coverage and passed through a quality filter as part of a data analysis pipeline, including sub-passaged replicates defined by a few SNPs, they can be accurately placed in a phylogenetic context. This reproducibility applies to all levels within and between serovars of Salmonella suggesting that investigators using these methods can have confidence in their conclusions.
下一代测序(NGS)越来越多地被用作一种分子流行病学工具,用于辨别最复杂、难以解决的细菌病原体的祖先和溯源。将可能的食物来源与临床分离株联系起来,需要将可疑病原体与环境背景区分开来,并将观察到的变异置于同一血清型内和其他密切相关的食源性病原体中发生的变异的更广泛背景下。同样重要的是,需要验证这些用于分子流行病学溯源的高分辨率分子工具。这些努力包括检查菌株聚类的稳定性以及亚培养对这些聚类的累积遗传效应。对包括不同谱系代表和许多重复克隆在内的大量 S. Montevideo 分离株进行了鸟枪法测序,以确定有多少变异是由于偏差、测序错误以及或分离株的培养造成的。所有新的草案基因组都与之前在基于 NGS 的分子流行病学案例研究中发表的 34 株 S. Montevideo 分离株进行了比较。
S. Montevideo 的血清内谱系差异数千个 SNP,仅略低于 S. Montevideo 与其他不同血清型之间观察到的 SNP 数量。在最近一次食源性暴发中涉及的单个 S. Montevideo 分支以及单个 NGS 重复中发现的变异较少。这些发现与先前记录罗氏 454 GS Titanium 技术中同聚物和缺失错误率的报告相似。然而,在任何情况下,与测序方法或样品制备相关的变异都不会导致与我们当前的系统发育结果或从这些数据中获得的随后的分子流行病学证据不一致。
实施经过验证的 NGS 数据采集和分析管道可提供高度可重复的结果,这些结果对于分子流行病学应用是稳定且可预测的。当以 15×-20×覆盖度收集草案基因组并作为数据分析管道的一部分通过质量过滤器(包括通过少数 SNP 定义的亚培养重复)时,它们可以准确地置于系统发育背景中。这种可重复性适用于沙门氏菌血清型内和血清型之间的所有级别,表明使用这些方法的研究人员可以对其结论充满信心。