Department of Human Genetics, Wellcome Trust Sanger Institute, Hinxton, UK.
Eur J Hum Genet. 2012 Jun;20(6):709-12. doi: 10.1038/ejhg.2011.274. Epub 2012 Feb 1.
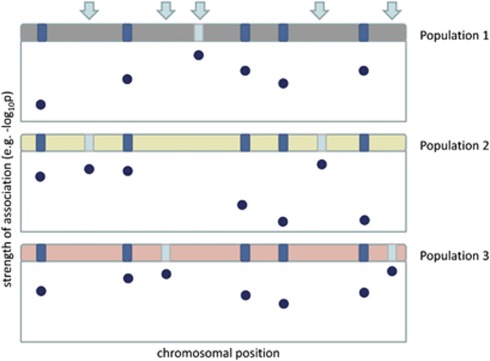
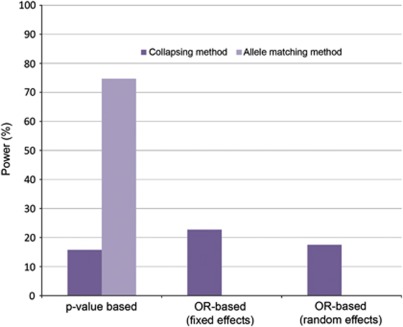
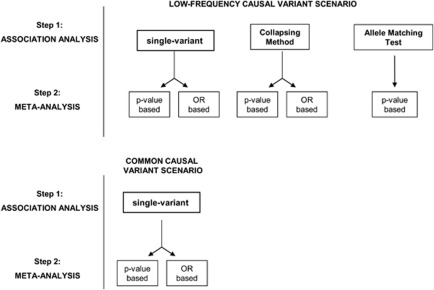
Meta-analysis has proven a useful tool in genetic association studies. Allelic heterogeneity can arise from ethnic background differences across populations being meta-analyzed (for example, in search of common frequency variants through genome-wide association studies), and through the presence of multiple low frequency and rare associated variants in the same functional unit of interest (for example, within a gene or a regulatory region). The latter challenge will be increasingly relevant in whole-genome and whole-exome sequencing studies investigating association with complex traits. Here, we evaluate the performance of different approaches to meta-analysis in the presence of allelic heterogeneity. We simulate allelic heterogeneity scenarios in three populations and examine the performance of current approaches to the analysis of these data. We show that current approaches can detect only a small fraction of common frequency causal variants. We also find that for low-frequency variants with large effects (odds ratios 2-3), single-point tests have high power, but also high false-positive rates. P-value based meta-analysis of summary results from allele-matching locus-wide tests outperforms collapsing approaches. We conclude that current strategies for the combination of genetic association data in the presence of allelic heterogeneity are insufficiently powered.
荟萃分析已被证明是遗传关联研究中的有用工具。等位基因异质性可能源于被荟萃分析的人群之间的种族背景差异(例如,通过全基因组关联研究寻找常见的频率变异),也可能源于同一功能单元中存在多个低频和罕见的相关变异(例如,在一个基因或调控区域内)。在全基因组和外显子组测序研究中,通过关联复杂性状来调查这些变异的作用,这种挑战将变得越来越重要。在这里,我们评估了在等位基因异质性存在的情况下,不同荟萃分析方法的性能。我们在三个群体中模拟了等位基因异质性的场景,并检查了当前分析这些数据的方法的性能。我们表明,当前的方法只能检测到一小部分常见的频率因果变异。我们还发现,对于具有大效应(比值比 2-3)的低频变异,单点检验具有高功效,但也具有高假阳性率。基于等位基因匹配的全基因组检验汇总结果的 P 值荟萃分析优于合并方法。我们得出的结论是,目前在存在等位基因异质性的情况下结合遗传关联数据的策略的功效不足。