Computer Science Department, University of British Columbia, Vancouver, BC, Canada.
BMC Bioinformatics. 2012 Feb 1;13:22. doi: 10.1186/1471-2105-13-22.
RNA molecules play critical roles in the cells of organisms, including roles in gene regulation, catalysis, and synthesis of proteins. Since RNA function depends in large part on its folded structures, much effort has been invested in developing accurate methods for prediction of RNA secondary structure from the base sequence. Minimum free energy (MFE) predictions are widely used, based on nearest neighbor thermodynamic parameters of Mathews, Turner et al. or those of Andronescu et al. Some recently proposed alternatives that leverage partition function calculations find the structure with maximum expected accuracy (MEA) or pseudo-expected accuracy (pseudo-MEA) methods. Advances in prediction methods are typically benchmarked using sensitivity, positive predictive value and their harmonic mean, namely F-measure, on datasets of known reference structures. Since such benchmarks document progress in improving accuracy of computational prediction methods, it is important to understand how measures of accuracy vary as a function of the reference datasets and whether advances in algorithms or thermodynamic parameters yield statistically significant improvements. Our work advances such understanding for the MFE and (pseudo-)MEA-based methods, with respect to the latest datasets and energy parameters.
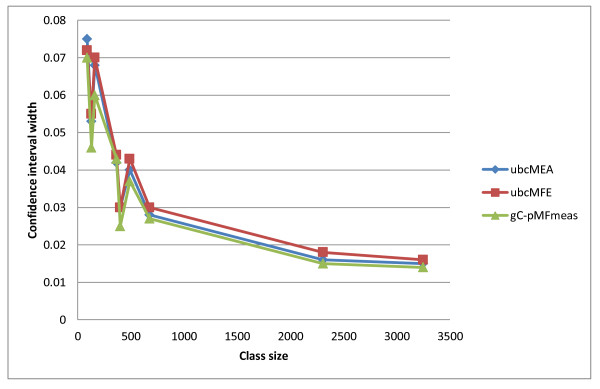
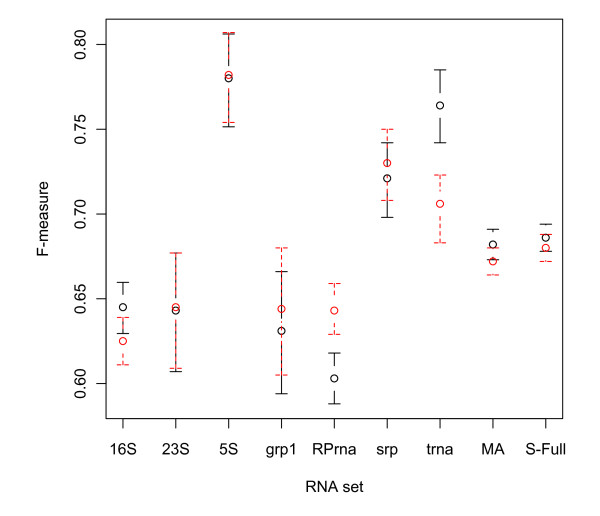
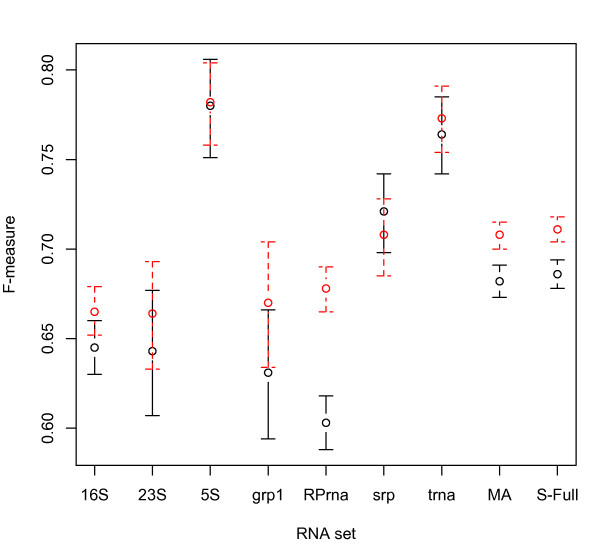
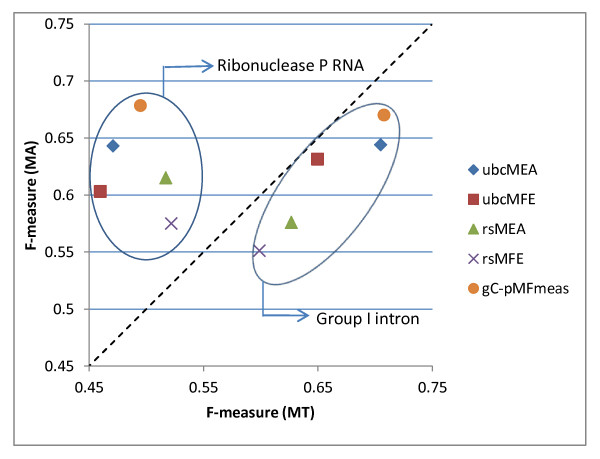
We present three main findings. First, using the bootstrap percentile method, we show that the average F-measure accuracy of the MFE and (pseudo-)MEA-based algorithms, as measured on our largest datasets with over 2000 RNAs from diverse families, is a reliable estimate (within a 2% range with high confidence) of the accuracy of a population of RNA molecules represented by this set. However, average accuracy on smaller classes of RNAs such as a class of 89 Group I introns used previously in benchmarking algorithm accuracy is not reliable enough to draw meaningful conclusions about the relative merits of the MFE and MEA-based algorithms. Second, on our large datasets, the algorithm with best overall accuracy is a pseudo MEA-based algorithm of Hamada et al. that uses a generalized centroid estimator of base pairs. However, between MFE and other MEA-based methods, there is no clear winner in the sense that the relative accuracy of the MFE versus MEA-based algorithms changes depending on the underlying energy parameters. Third, of the four parameter sets we considered, the best accuracy for the MFE-, MEA-based, and pseudo-MEA-based methods is 0.686, 0.680, and 0.711, respectively (on a scale from 0 to 1 with 1 meaning perfect structure predictions) and is obtained with a thermodynamic parameter set obtained by Andronescu et al. called BL* (named after the Boltzmann likelihood method by which the parameters were derived).
Large datasets should be used to obtain reliable measures of the accuracy of RNA structure prediction algorithms, and average accuracies on specific classes (such as Group I introns and Transfer RNAs) should be interpreted with caution, considering the relatively small size of currently available datasets for such classes. The accuracy of the MEA-based methods is significantly higher when using the BL* parameter set of Andronescu et al. than when using the parameters of Mathews and Turner, and there is no significant difference between the accuracy of MEA-based methods and MFE when using the BL* parameters. The pseudo-MEA-based method of Hamada et al. with the BL* parameter set significantly outperforms all other MFE and MEA-based algorithms on our large data sets.
RNA 分子在生物细胞中发挥着关键作用,包括在基因调控、催化和蛋白质合成中的作用。由于 RNA 的功能在很大程度上取决于其折叠结构,因此人们投入了大量精力来开发从碱基序列预测 RNA 二级结构的准确方法。最小自由能 (MFE) 预测是基于 Mathews、Turner 等人的最近邻热力学参数或 Andronescu 等人的参数广泛使用的。一些最近提出的利用分区函数计算的替代方法找到了具有最大预期准确性 (MEA) 或伪预期准确性 (pseudo-MEA) 的结构。预测方法的进展通常使用在具有已知参考结构的数据集上的灵敏度、阳性预测值及其调和平均值(即 F 度量)进行基准测试。由于此类基准测试记录了计算预测方法准确性提高的进展,因此了解准确性度量如何随参考数据集变化以及算法或热力学参数的改进是否产生统计学上显著的改进非常重要。我们的工作针对最新的数据集和能量参数,针对 MFE 和(伪)MEA 方法,推进了这种理解。
我们提出了三个主要发现。首先,使用自举百分位法,我们表明,基于 MFE 和(伪)MEA 的算法的平均 F 度量准确性,在我们最大的数据集上进行测量,该数据集包含来自多个家族的 2000 多个 RNA,是该数据集所代表的 RNA 分子群体的可靠估计(在置信度高的情况下,范围在 2% 以内)。然而,在较小的 RNA 类(如以前用于基准算法准确性的 89 个 I 类内含子类)上的平均准确性不够可靠,无法对 MFE 和基于 MEA 的算法的相对优点得出有意义的结论。其次,在我们的大型数据集上,整体准确性最好的算法是 Hamada 等人的基于伪 MEA 的算法,该算法使用碱基对的广义质心估计器。然而,在 MFE 和其他基于 MEA 的方法之间,没有明显的赢家,因为 MFE 与基于 MEA 的算法的相对准确性取决于基础能量参数。第三,在我们考虑的四个参数集中,MFE、MEA 基于和基于 pseudo-MEA 的方法的最佳准确性分别为 0.686、0.680 和 0.711(在 0 到 1 的范围内,1 表示完美的结构预测),并且使用由 Andronescu 等人提出的称为 BL*的热力学参数集(以推导参数的玻尔兹曼似然法命名)获得。
应该使用大型数据集来获得 RNA 结构预测算法准确性的可靠度量,并且应该谨慎解释特定类(如 I 类内含子和转移 RNA)的平均准确性,因为目前此类类别的数据集相对较小。当使用 Andronescu 等人的 BL参数集时,基于 MEA 的方法的准确性明显高于使用 Mathews 和 Turner 的参数时,并且当使用 BL参数时,基于 MEA 的方法和 MFE 的准确性之间没有显著差异。Hamada 等人的基于伪 MEA 的方法使用 BL*参数集在我们的大型数据集上明显优于所有其他基于 MFE 和基于 MEA 的算法。