Key Laboratory of Aquatic Biodiversity and Conservation, Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, People's Republic of China.
PLoS One. 2012;7(2):e30630. doi: 10.1371/journal.pone.0030630. Epub 2012 Feb 7.
The ciliated protozoan Tetrahymena thermophila is a well-studied single-celled eukaryote model organism for cellular and molecular biology. However, the lack of extensive T. thermophila cDNA libraries or a large expressed sequence tag (EST) database limited the quality of the original genome annotation.
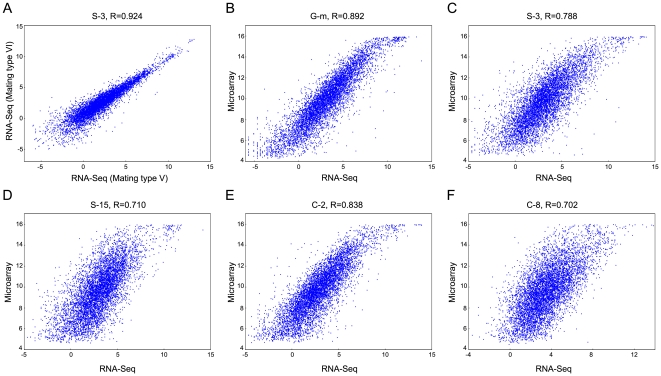
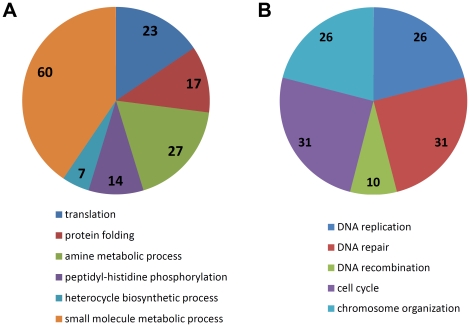
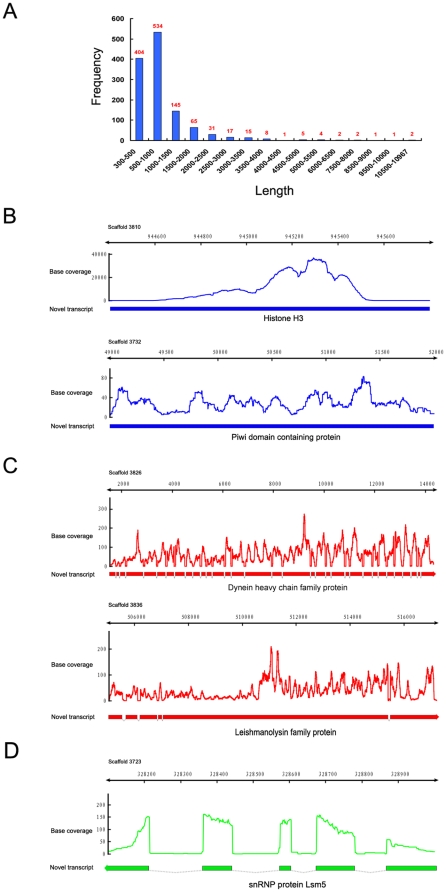
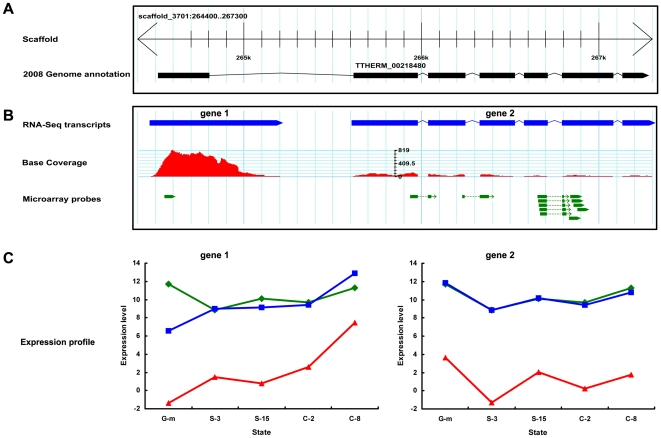
METHODOLOGY/PRINCIPAL FINDINGS: This RNA-seq study describes the first deep sequencing analysis of the T. thermophila transcriptome during the three major stages of the life cycle: growth, starvation and conjugation. Uniquely mapped reads covered more than 96% of the 24,725 predicted gene models in the somatic genome. More than 1,000 new transcribed regions were identified. The great dynamic range of RNA-seq allowed detection of a nearly six order-of-magnitude range of measurable gene expression orchestrated by this cell. RNA-seq also allowed the first prediction of transcript untranslated regions (UTRs) and an updated (larger) size estimate of the T. thermophila transcriptome: 57 Mb, or about 55% of the somatic genome. Our study identified nearly 1,500 alternative splicing (AS) events distributed over 5.2% of T. thermophila genes. This percentage represents a two order-of-magnitude increase over previous EST-based estimates in Tetrahymena. Evidence of stage-specific regulation of alternative splicing was also obtained. Finally, our study allowed us to completely confirm about 26.8% of the genes originally predicted by the gene finder, to correct coding sequence boundaries and intron-exon junctions for about a third, and to reassign microarray probes and correct earlier microarray data.
CONCLUSIONS/SIGNIFICANCE: RNA-seq data significantly improve the genome annotation and provide a fully comprehensive view of the global transcriptome of T. thermophila. To our knowledge, 5.2% of T. thermophila genes with AS is the highest percentage of genes showing AS reported in a unicellular eukaryote. Tetrahymena thus becomes an excellent unicellular model eukaryote in which to investigate mechanisms of alternative splicing.
纤毛原生动物嗜热四膜虫是一种研究较为深入的单细胞真核生物模式生物,可用于细胞和分子生物学研究。然而,由于缺乏广泛的嗜热四膜虫 cDNA 文库或大型表达序列标签 (EST) 数据库,限制了原始基因组注释的质量。
方法/主要发现:本 RNA-seq 研究描述了嗜热四膜虫转录组在生命周期的三个主要阶段(生长、饥饿和接合)中的首次深度测序分析。唯一映射的读数覆盖了 24725 个预测的体细胞基因模型中的 96%以上。鉴定出 1000 多个新的转录区域。RNA-seq 的大动态范围允许检测到由该细胞协调的可测量基因表达的近六个数量级范围。RNA-seq 还首次预测了转录物非翻译区 (UTR),并更新了(更大)嗜热四膜虫转录组的大小估计:57Mb,约占体细胞基因组的 55%。我们的研究鉴定了近 1500 个分布在 5.2%嗜热四膜虫基因中的选择性剪接 (AS) 事件。这一百分比代表了 Tetrahymena 中以前基于 EST 的估计值的两个数量级的增加。还获得了选择性剪接的阶段特异性调节的证据。最后,我们的研究使我们能够完全确认基因预测者最初预测的约 26.8%的基因,修正了大约三分之一的编码序列边界和内含子-外显子接头,并重新分配了微阵列探针和修正了早期的微阵列数据。
结论/意义:RNA-seq 数据显著提高了基因组注释,并提供了嗜热四膜虫全转录组的全面视图。据我们所知,嗜热四膜虫中具有 AS 的基因的 5.2%是在单细胞真核生物中报道的具有 AS 的基因的最高百分比。因此,嗜热四膜虫成为研究选择性剪接机制的优秀单细胞真核模式生物。