Miao Wei, Xiong Jie, Bowen Josephine, Wang Wei, Liu Yifan, Braguinets Olga, Grigull Jorg, Pearlman Ronald E, Orias Eduardo, Gorovsky Martin A
Department of Biology, University of Rochester, Rochester, New York, USA.
PLoS One. 2009;4(2):e4429. doi: 10.1371/journal.pone.0004429. Epub 2009 Feb 10.
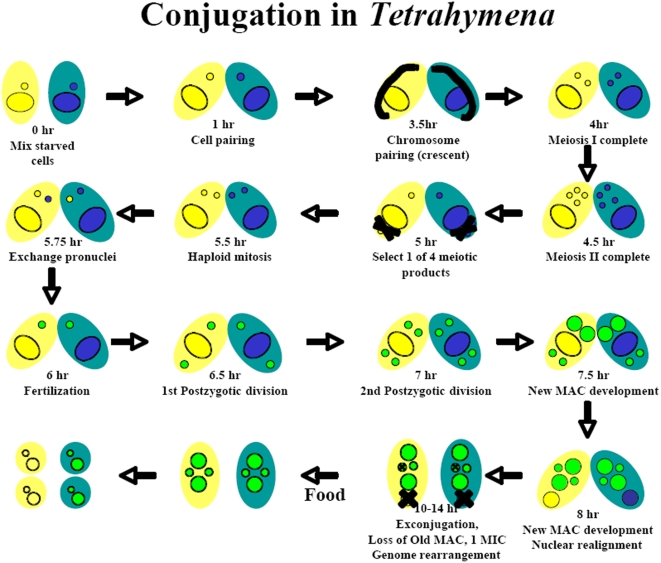
The model eukaryote, Tetrahymena thermophila, is the first ciliated protozoan whose genome has been sequenced, enabling genome-wide analysis of gene expression.
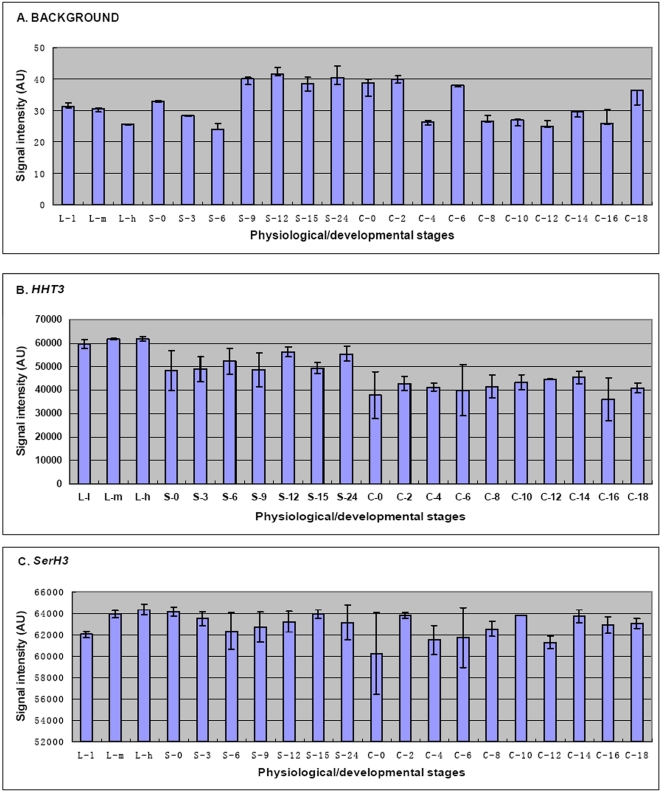
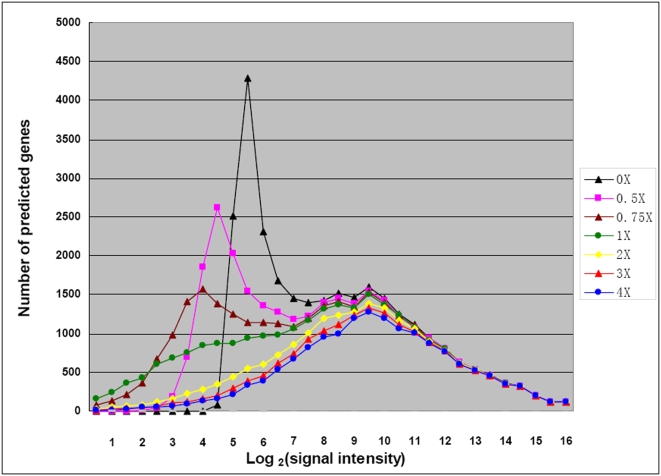
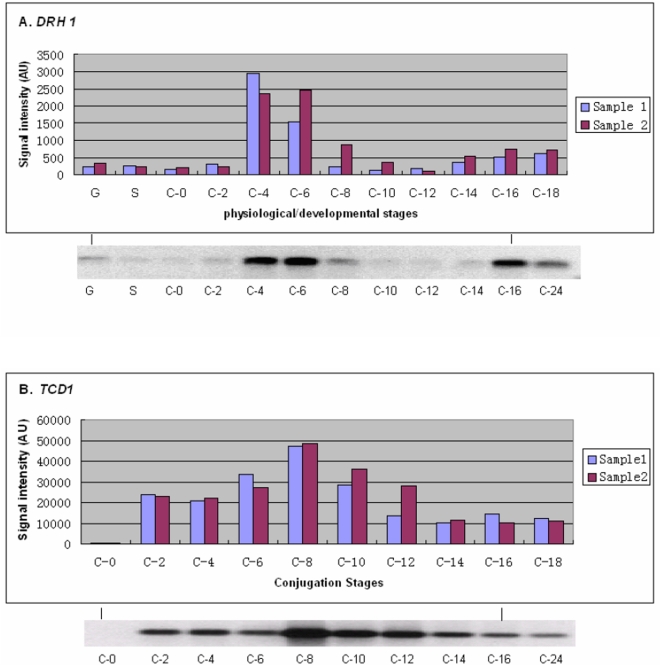
METHODOLOGY/PRINCIPAL FINDINGS: A genome-wide microarray platform containing the predicted coding sequences (putative genes) for T. thermophila is described, validated and used to study gene expression during the three major stages of the organism's life cycle: growth, starvation and conjugation.
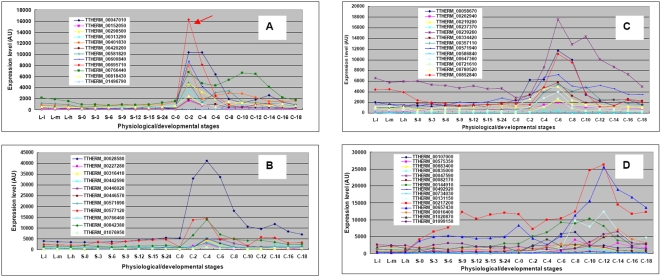
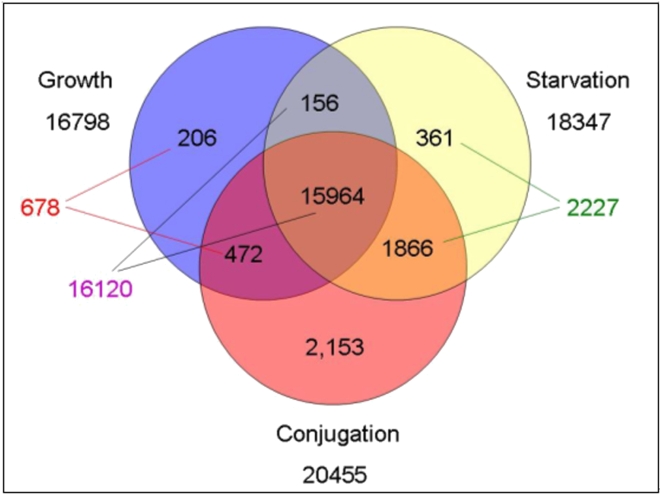
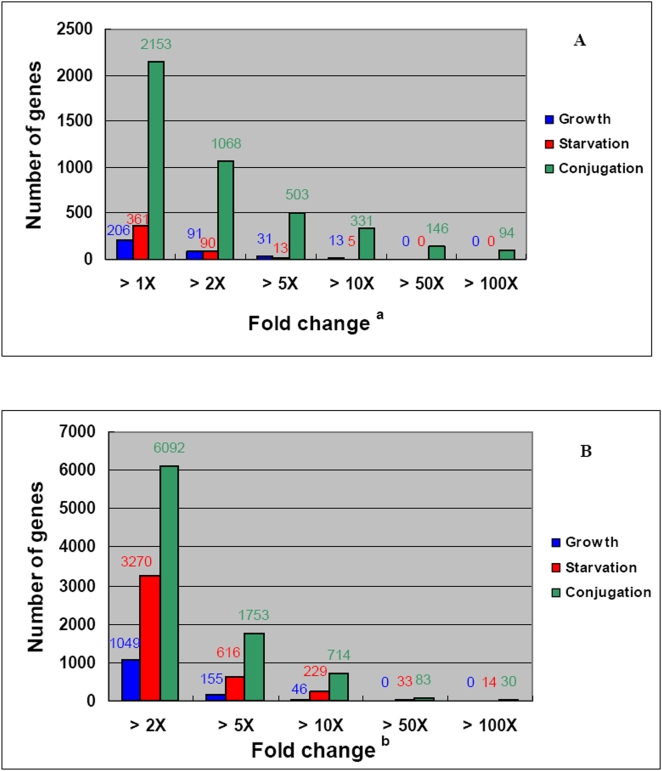
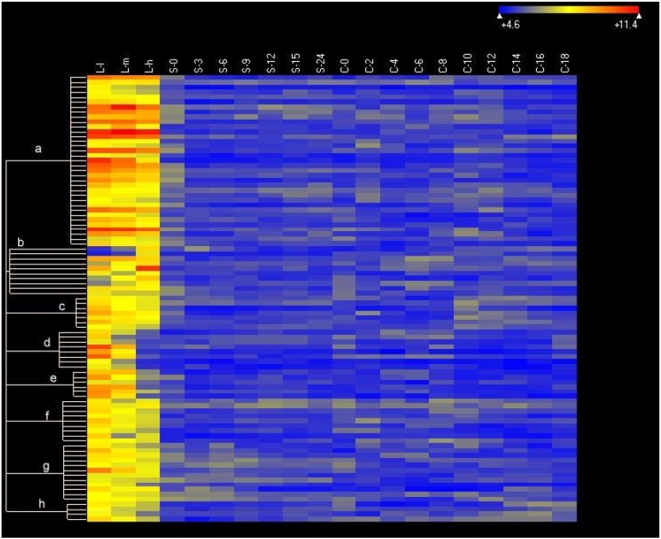
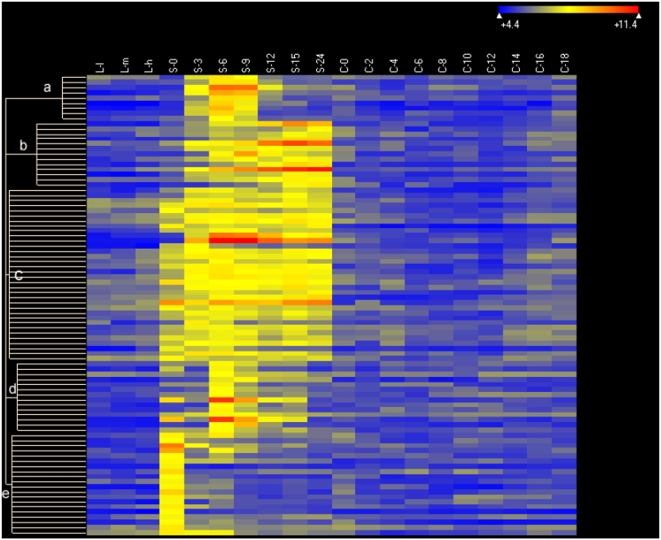
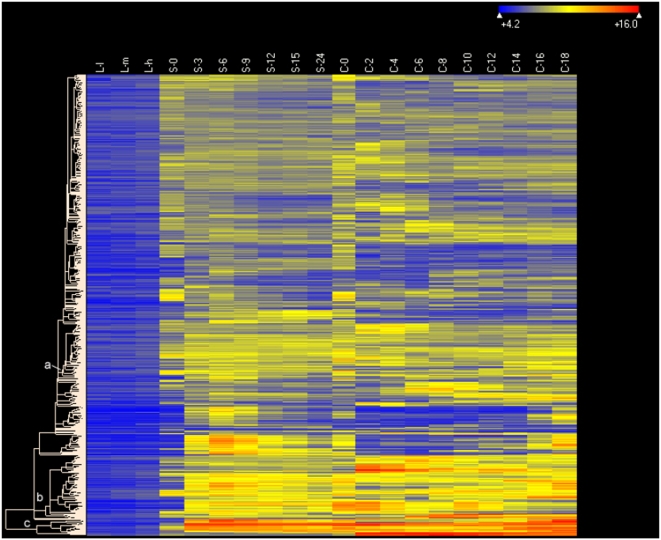
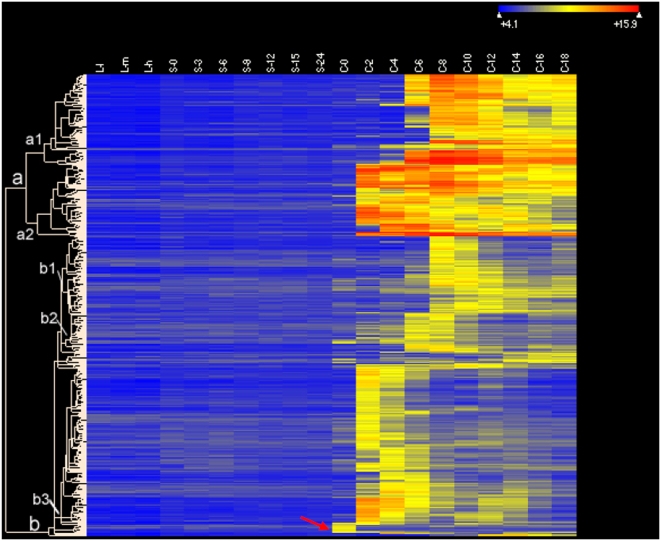
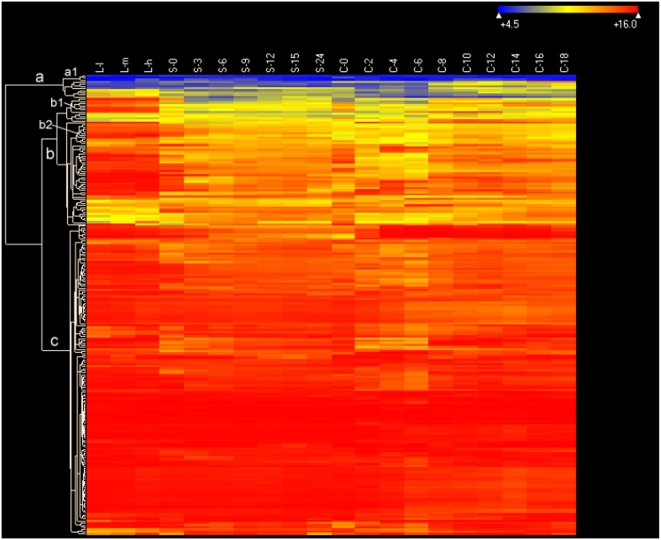
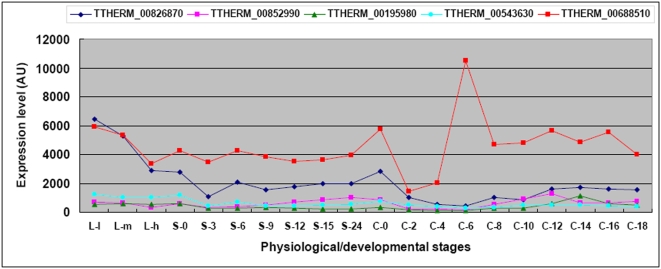
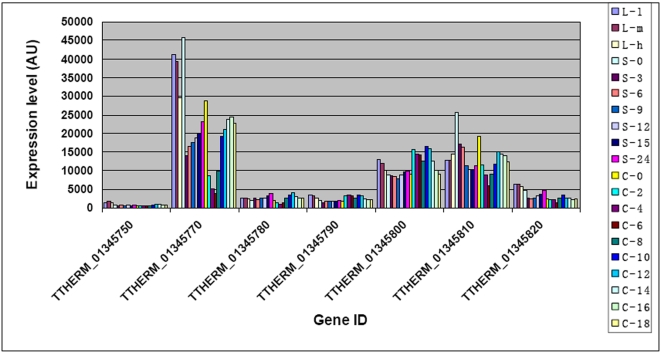
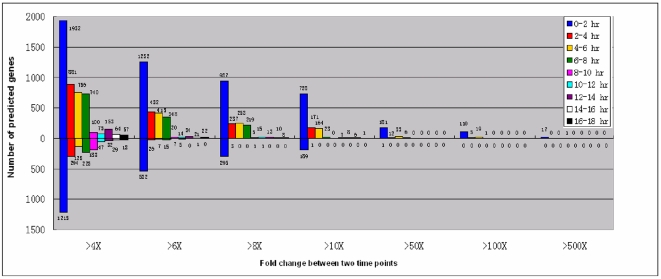
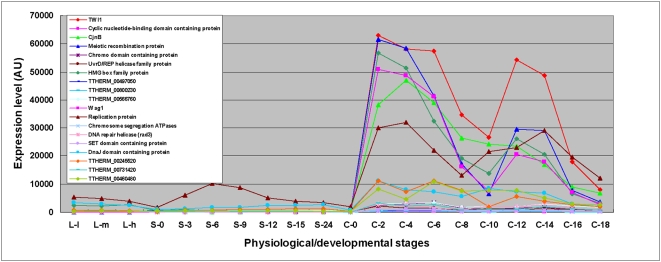
CONCLUSIONS/SIGNIFICANCE: Of the approximately 27,000 predicted open reading frames, transcripts homologous to only approximately 5900 are not detectable in any of these life cycle stages, indicating that this single-celled organism does indeed contain a large number of functional genes. Transcripts from over 5000 predicted genes are expressed at levels >5x corrected background and 95 genes are expressed at >250x corrected background in all stages. Transcripts homologous to 91 predicted genes are specifically expressed and 155 more are highly up-regulated in growing cells, while 90 are specifically expressed and 616 are up-regulated during starvation. Strikingly, transcripts homologous to 1068 predicted genes are specifically expressed and 1753 are significantly up-regulated during conjugation. The patterns of gene expression during conjugation correlate well with the developmental stages of meiosis, nuclear differentiation and DNA elimination. The relationship between gene expression and chromosome fragmentation is analyzed. Genes encoding proteins known to interact or to function in complexes show similar expression patterns, indicating that co-ordinate expression with putative genes of known function can identify genes with related functions. New candidate genes associated with the RNAi-like process of DNA elimination and with meiosis are identified and the late stages of conjugation are shown to be characterized by specific expression of an unexpectedly large and diverse number of genes not involved in nuclear functions.
模式真核生物嗜热四膜虫是首个基因组已被测序的纤毛原生动物,这使得对基因表达进行全基因组分析成为可能。
方法/主要发现:本文描述、验证了一个包含嗜热四膜虫预测编码序列(推定基因)的全基因组微阵列平台,并将其用于研究该生物体生命周期三个主要阶段(生长、饥饿和接合)中的基因表达。
结论/意义:在大约27000个预测的开放阅读框中,在这些生命周期阶段的任何一个中都检测不到与仅约5900个同源的转录本,这表明这种单细胞生物确实含有大量功能基因。超过5000个预测基因的转录本在所有阶段的表达水平都高于5倍校正背景,95个基因在所有阶段的表达水平高于250倍校正背景。与91个预测基因同源的转录本在生长细胞中特异性表达,另外155个高度上调,而90个在饥饿期间特异性表达,616个上调。引人注目的是,与1068个预测基因同源的转录本在接合期间特异性表达,1753个显著上调。接合期间的基因表达模式与减数分裂、核分化和DNA消除的发育阶段密切相关。分析了基因表达与染色体片段化之间的关系。编码已知在复合物中相互作用或起作用的蛋白质的基因表现出相似的表达模式,这表明与已知功能的推定基因协同表达可以鉴定具有相关功能的基因。鉴定了与DNA消除的RNAi样过程和减数分裂相关的新候选基因,并且接合后期的特征是大量未参与核功能的基因意外地特异性表达且种类多样。