Department of Medical Statistics, University of Göttingen, Göttingen, Germany.
PLoS One. 2012;7(2):e31242. doi: 10.1371/journal.pone.0031242. Epub 2012 Feb 21.
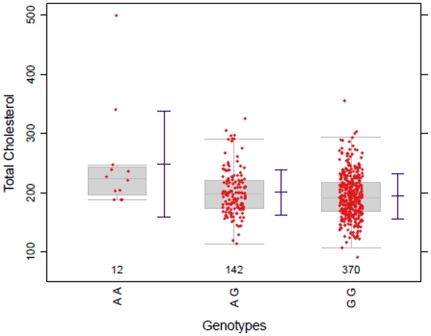
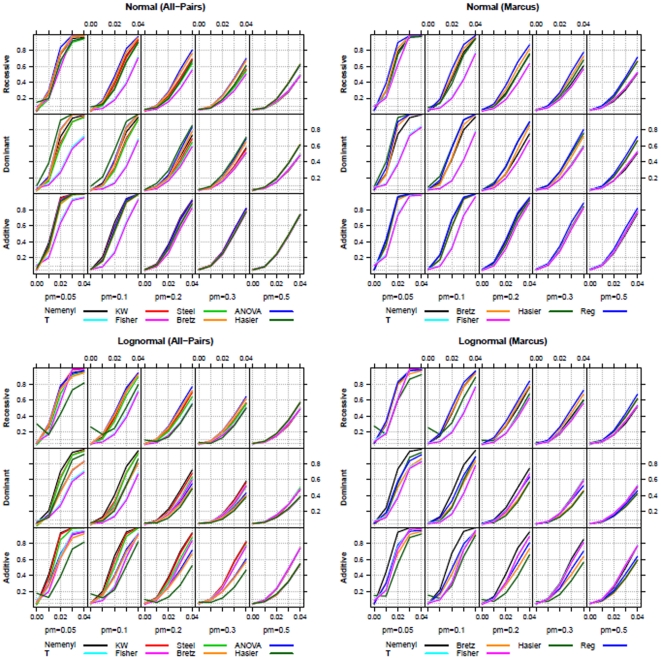
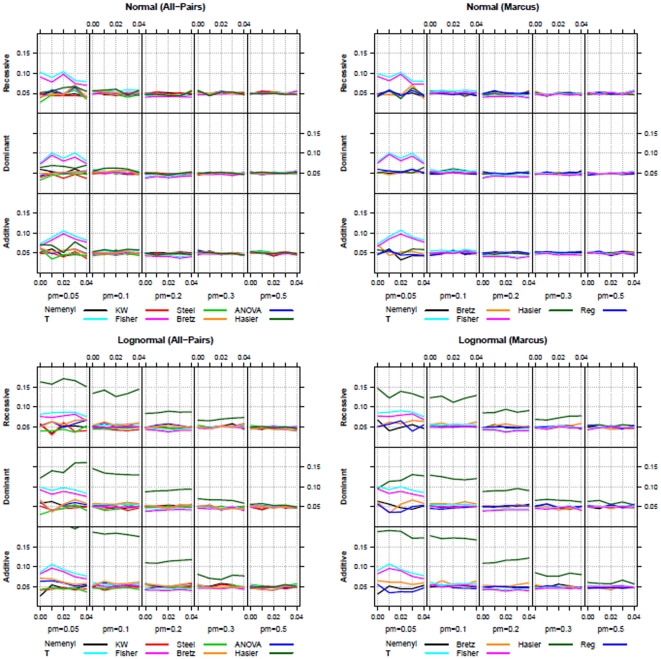
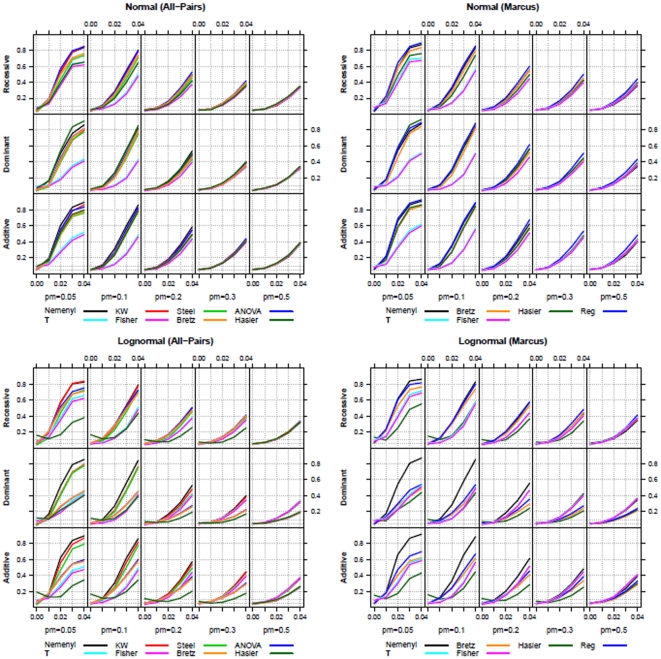
Statistical association between a single nucleotide polymorphism (SNP) genotype and a quantitative trait in genome-wide association studies is usually assessed using a linear regression model, or, in the case of non-normally distributed trait values, using the Kruskal-Wallis test. While linear regression models assume an additive mode of inheritance via equi-distant genotype scores, Kruskal-Wallis test merely tests global differences in trait values associated with the three genotype groups. Both approaches thus exhibit suboptimal power when the underlying inheritance mode is dominant or recessive. Furthermore, these tests do not perform well in the common situations when only a few trait values are available in a rare genotype category (disbalance), or when the values associated with the three genotype categories exhibit unequal variance (variance heterogeneity). We propose a maximum test based on Marcus-type multiple contrast test for relative effect sizes. This test allows model-specific testing of either dominant, additive or recessive mode of inheritance, and it is robust against variance heterogeneity. We show how to obtain mode-specific simultaneous confidence intervals for the relative effect sizes to aid in interpreting the biological relevance of the results. Further, we discuss the use of a related all-pairwise comparisons contrast test with range preserving confidence intervals as an alternative to Kruskal-Wallis heterogeneity test. We applied the proposed maximum test to the Bogalusa Heart Study dataset, and gained a remarkable increase in the power to detect association, particularly for rare genotypes. Our simulation study also demonstrated that the proposed non-parametric tests control family-wise error rate in the presence of non-normality and variance heterogeneity contrary to the standard parametric approaches. We provide a publicly available R library nparcomp that can be used to estimate simultaneous confidence intervals or compatible multiplicity-adjusted p-values associated with the proposed maximum test.
在全基因组关联研究中,通常使用线性回归模型评估单核苷酸多态性 (SNP) 基因型与定量性状之间的统计关联,或者在性状值呈非正态分布的情况下,使用 Kruskal-Wallis 检验。虽然线性回归模型假设通过等距基因型得分的加性遗传模式,但 Kruskal-Wallis 检验仅检验与三种基因型组相关的性状值的全局差异。当潜在的遗传模式为显性或隐性时,这两种方法的功效都不理想。此外,当稀有基因型类别中只有少数性状值可用(不平衡),或者与三种基因型类别相关的性状值表现出不等方差(方差异质性)时,这些检验也不能很好地发挥作用。我们提出了一种基于马库斯型多重对比检验的最大检验,用于相对效应大小。该检验允许针对显性、加性或隐性遗传模式进行特定于模型的检验,并且对方差异质性具有稳健性。我们展示了如何获得特定于模式的相对效应大小的同时置信区间,以帮助解释结果的生物学相关性。此外,我们讨论了使用具有保留范围置信区间的相关全对比较对比检验作为 Kruskal-Wallis 异质性检验的替代方法。我们将提出的最大检验应用于 Bogalusa 心脏研究数据集,并显著提高了检测关联的功效,特别是对于稀有基因型。我们的模拟研究还表明,与标准参数方法相反,所提出的非参数检验在非正态性和方差异质性存在的情况下控制了家族错误率。我们提供了一个公共可用的 R 库 nparcomp,可用于估计与提出的最大检验相关的同时置信区间或兼容的多重调整 p 值。