Dementia Research Centre, Department of Neurodegenerative Diseases, UCL Institute of Neurology, London WC1N 3BG, UK.
Brain. 2012 Mar;135(Pt 3):736-50. doi: 10.1093/brain/awr361.
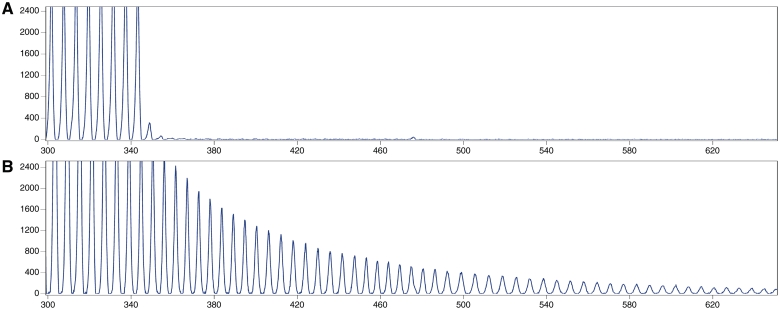
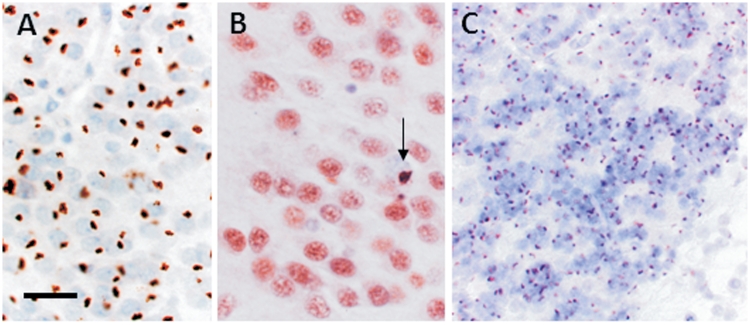
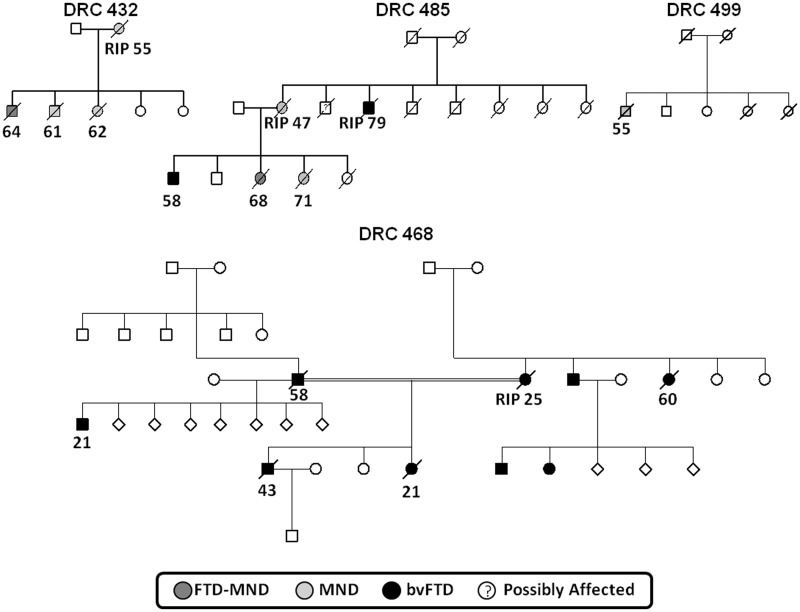
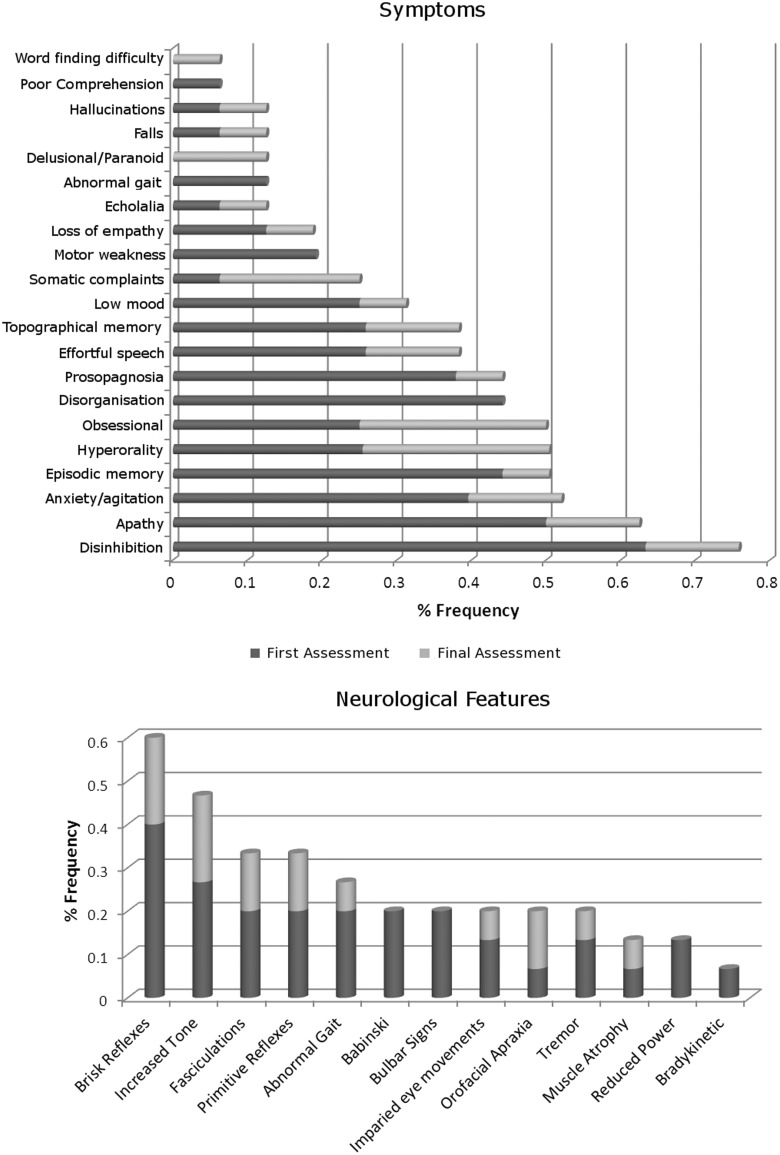
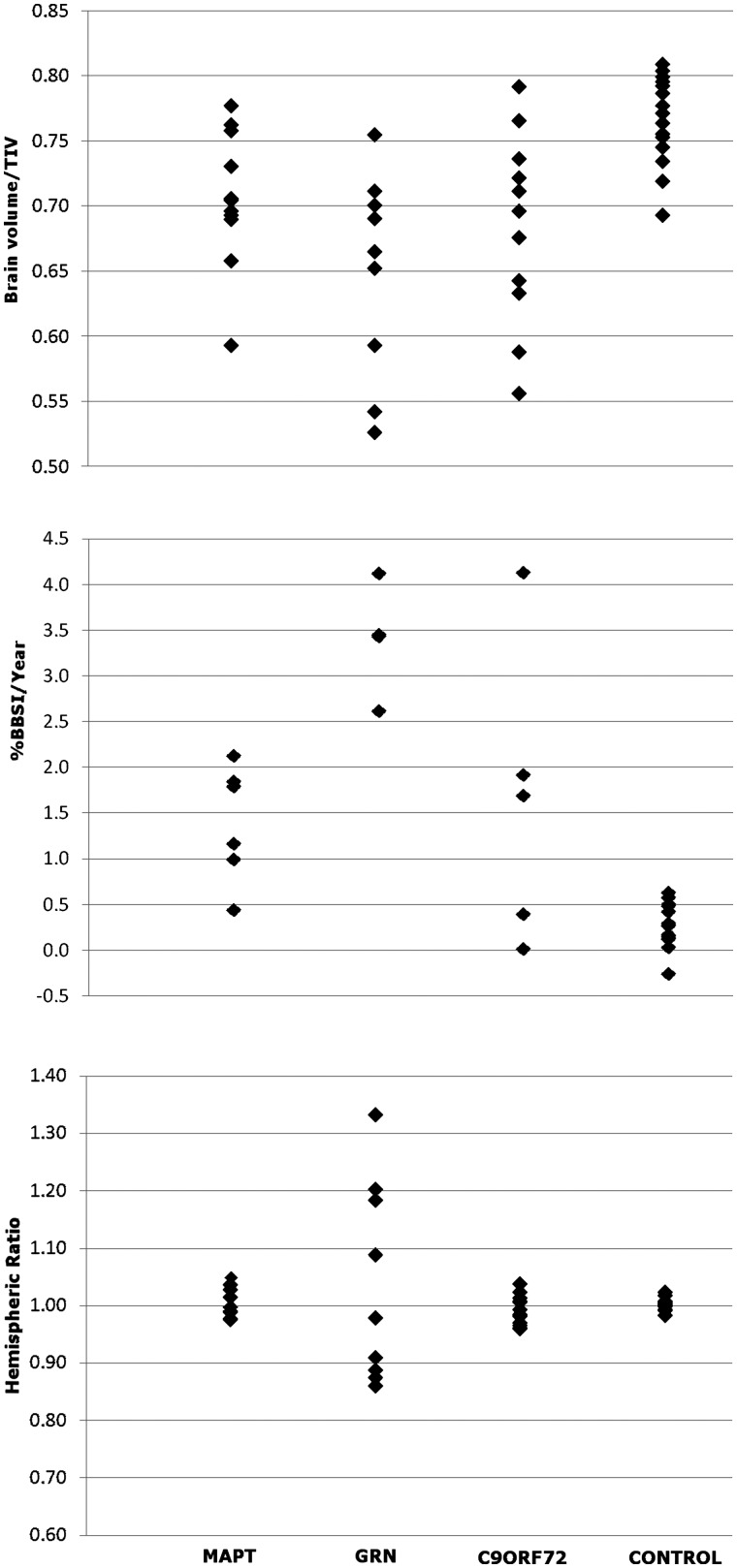
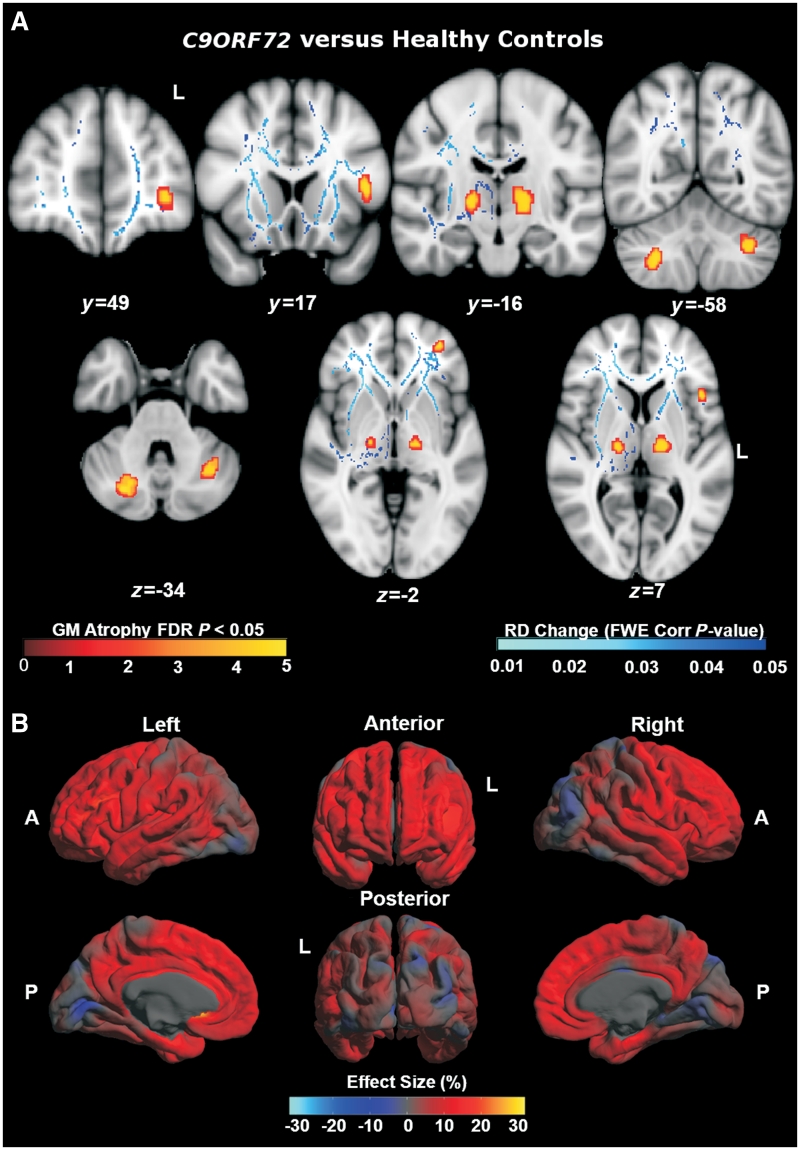
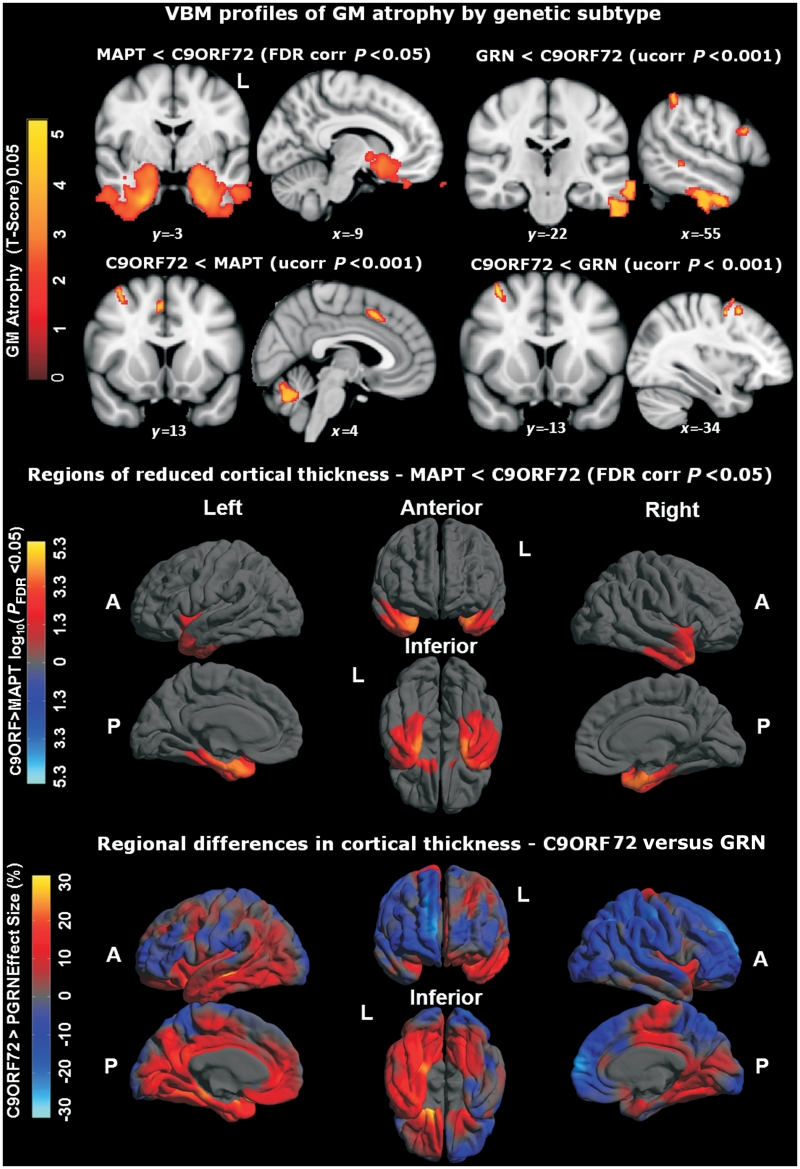
An expanded hexanucleotide repeat in the C9ORF72 gene has recently been identified as a major cause of familial frontotemporal lobar degeneration and motor neuron disease, including cases previously identified as linked to chromosome 9. Here we present a detailed retrospective clinical, neuroimaging and histopathological analysis of a C9ORF72 mutation case series in relation to other forms of genetically determined frontotemporal lobar degeneration ascertained at a specialist centre. Eighteen probands (19 cases in total) were identified, representing 35% of frontotemporal lobar degeneration cases with identified mutations, 36% of cases with clinical evidence of motor neuron disease and 7% of the entire cohort. Thirty-three per cent of these C9ORF72 cases had no identified relevant family history. Families showed wide variation in clinical onset (43-68 years) and duration (1.7-22 years). The most common presenting syndrome (comprising a half of cases) was behavioural variant frontotemporal dementia, however, there was substantial clinical heterogeneity across the C9ORF72 mutation cohort. Sixty per cent of cases developed clinical features consistent with motor neuron disease during the period of follow-up. Anxiety and agitation and memory impairment were prominent features (between a half to two-thirds of cases), and dominant parietal dysfunction was also frequent. Affected individuals showed variable magnetic resonance imaging findings; however, relative to healthy controls, the group as a whole showed extensive thinning of frontal, temporal and parietal cortices, subcortical grey matter atrophy including thalamus and cerebellum and involvement of long intrahemispheric, commissural and corticospinal tracts. The neuroimaging profile of the C9ORF72 expansion was significantly more symmetrical than progranulin mutations with significantly less temporal lobe involvement than microtubule-associated protein tau mutations. Neuropathological examination in six cases with C9ORF72 mutation from the frontotemporal lobar degeneration series identified histomorphological features consistent with either type A or B TAR DNA-binding protein-43 deposition; however, p62-positive (in excess of TAR DNA-binding protein-43 positive) neuronal cytoplasmic inclusions in hippocampus and cerebellum were a consistent feature of these cases, in contrast to the similar frequency of p62 and TAR DNA-binding protein-43 deposition in 53 control cases with frontotemporal lobar degeneration-TAR DNA-binding protein. These findings corroborate the clinical importance of the C9ORF72 mutation in frontotemporal lobar degeneration, delineate phenotypic and neuropathological features that could help to guide genetic testing, and suggest hypotheses for elucidating the neurobiology of a culprit subcortical network.
C9ORF72 基因中的六核苷酸重复扩增最近被确定为家族性额颞叶变性和运动神经元病的主要原因,包括以前被确定与 9 号染色体相关的病例。在这里,我们呈现了一个详细的回顾性临床、神经影像学和组织病理学分析,涉及在一个专门中心确定的其他形式的遗传性额颞叶变性病例系列。确定了 18 个先证者(共 19 例),占具有确定突变的额颞叶变性病例的 35%,具有临床证据的运动神经元病病例的 36%,以及整个队列的 7%。这些 C9ORF72 病例中有 33%没有确定的相关家族史。家族表现出广泛的临床发病年龄(43-68 岁)和病程(1.7-22 年)变化。这些 C9ORF72 病例中有 33%没有确定的相关家族史。家族表现出广泛的临床发病年龄(43-68 岁)和病程(1.7-22 年)变化。这些 C9ORF72 病例中有 33%没有确定的相关家族史。家族表现出广泛的临床发病年龄(43-68 岁)和病程(1.7-22 年)变化。家族表现出广泛的临床发病年龄(43-68 岁)和病程(1.7-22 年)变化。家族表现出广泛的临床发病年龄(43-68 岁)和病程(1.7-22 年)变化。
家族表现出广泛的临床发病年龄(43-68 岁)和病程(1.7-22 年)变化。
最常见的首发综合征(占一半病例)为行为变异型额颞叶痴呆,但在 C9ORF72 突变队列中存在明显的临床异质性。在随访期间,60%的病例出现符合运动神经元病的临床特征。焦虑和激越以及记忆障碍是突出的特征(占一半至三分之二的病例),并且显性顶叶功能障碍也很常见。受影响的个体表现出不同的磁共振成像发现;然而,与健康对照组相比,该组整体表现出额叶、颞叶和顶叶皮质广泛变薄,皮质下灰质萎缩包括丘脑和小脑,以及长内半球、连合和皮质脊髓束受累。C9ORF72 扩张的神经影像学特征明显比颗粒蛋白突变更对称,与微管相关蛋白 tau 突变相比,颞叶受累明显较少。从额颞叶变性系列中确定的六个 C9ORF72 突变病例的神经病理学检查确定了与 A 型或 B 型 TAR DNA 结合蛋白-43 沉积一致的组织形态学特征;然而,在这些病例中,海马和小脑中 p62 阳性(超过 TAR DNA 结合蛋白-43 阳性)神经元细胞质包含物是一个一致的特征,与在具有额颞叶变性-TAR DNA 结合蛋白的 53 个对照病例中 p62 和 TAR DNA 结合蛋白-43 沉积的相似频率形成对比。这些发现证实了 C9ORF72 突变在额颞叶变性中的临床重要性,描绘了有助于指导基因检测的表型和神经病理学特征,并提出了阐明潜在皮质下网络神经生物学的假设。