Department of Neurology, Mayo Clinic, Rochester, Minnesota 55905, USA.
Brain. 2012 Mar;135(Pt 3):765-83. doi: 10.1093/brain/aws004.
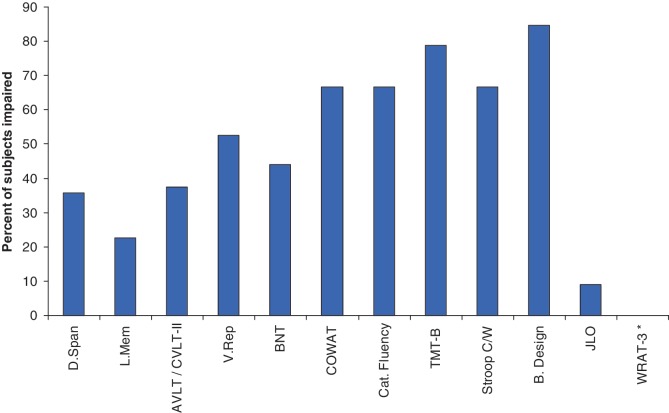
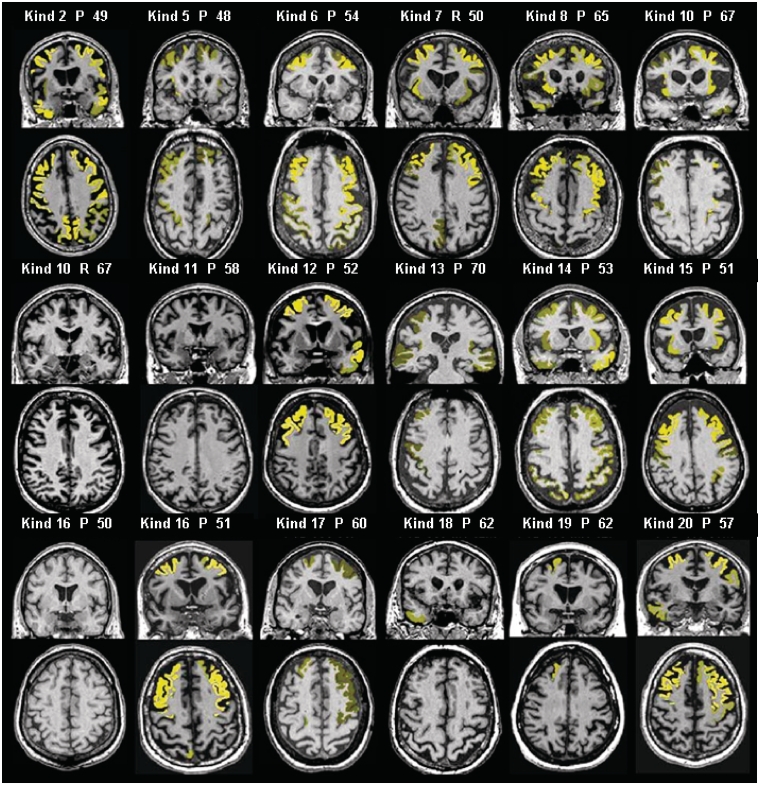
Numerous kindreds with familial frontotemporal dementia and/or amyotrophic lateral sclerosis have been linked to chromosome 9, and an expansion of the GGGGCC hexanucleotide repeat in the non-coding region of chromosome 9 open reading frame 72 has recently been identified as the pathogenic mechanism. We describe the key characteristics in the probands and their affected relatives who have been evaluated at Mayo Clinic Rochester or Mayo Clinic Florida in whom the hexanucleotide repeat expansion were found. Forty-three probands and 10 of their affected relatives with DNA available (total 53 subjects) were shown to carry the hexanucleotide repeat expansion. Thirty-six (84%) of the 43 probands had a familial disorder, whereas seven (16%) appeared to be sporadic. Among examined subjects from the 43 families (n = 63), the age of onset ranged from 33 to 72 years (median 52 years) and survival ranged from 1 to 17 years, with the age of onset <40 years in six (10%) and >60 in 19 (30%). Clinical diagnoses among examined subjects included behavioural variant frontotemporal dementia with or without parkinsonism (n = 30), amyotrophic lateral sclerosis (n = 18), frontotemporal dementia/amyotrophic lateral sclerosis with or without parkinsonism (n = 12), and other various syndromes (n = 3). Parkinsonism was present in 35% of examined subjects, all of whom had behavioural variant frontotemporal dementia or frontotemporal dementia/amyotrophic lateral sclerosis as the dominant clinical phenotype. No subject with a diagnosis of primary progressive aphasia was identified with this mutation. Incomplete penetrance was suggested in two kindreds, and the youngest generation had significantly earlier age of onset (>10 years) compared with the next oldest generation in 11 kindreds. Neuropsychological testing showed a profile of slowed processing speed, complex attention/executive dysfunction, and impairment in rapid word retrieval. Neuroimaging studies showed bilateral frontal abnormalities most consistently, with more variable degrees of parietal with or without temporal changes; no case had strikingly focal or asymmetric findings. Neuropathological examination of 14 patients revealed a range of transactive response DNA binding protein molecular weight 43 pathology (10 type A and four type B), as well as ubiquitin-positive cerebellar granular neuron inclusions in all but one case. Motor neuron degeneration was detected in nine patients, including five patients without ante-mortem signs of motor neuron disease. While variability exists, most cases with this mutation have a characteristic spectrum of demographic, clinical, neuropsychological, neuroimaging and especially neuropathological findings.
许多伴有家族性额颞叶痴呆和/或肌萎缩侧索硬化症的家系与 9 号染色体相关联,最近发现非编码区 9 号染色体开放阅读框 72 的 GGGGCC 六核苷酸重复扩展是致病机制。我们描述了在梅奥诊所罗切斯特或梅奥诊所佛罗里达接受评估的先证者及其受影响亲属的关键特征,这些先证者及其亲属的六核苷酸重复扩展已被发现。43 名先证者及其 10 名携带 DNA 的受影响亲属(共 53 名受试者)被证实携带六核苷酸重复扩展。36 名(84%)先证者患有家族性疾病,而 7 名(16%)似乎为散发性。在 43 个家族的受检者中(n=63),发病年龄从 33 岁到 72 岁不等(中位年龄 52 岁),存活时间从 1 年到 17 年不等,其中 6 名(10%)发病年龄<40 岁,19 名(30%)发病年龄>60 岁。受检者的临床诊断包括伴有或不伴有帕金森病的行为变异型额颞叶痴呆(n=30)、肌萎缩侧索硬化症(n=18)、额颞叶痴呆/肌萎缩侧索硬化症伴有或不伴有帕金森病(n=12)以及其他各种综合征(n=3)。35%的受检者存在帕金森病,所有这些患者均为行为变异型额颞叶痴呆或额颞叶痴呆/肌萎缩侧索硬化症作为主要的临床表型。未发现携带该突变的原发性进行性失语症患者。两个家系提示不完全外显,11 个家系中最年轻的一代与次年轻的一代相比,发病年龄明显提前(>10 年)。神经心理学测试显示出处理速度减慢、复杂注意力/执行功能障碍和快速单词检索受损的特征。神经影像学研究最一致地显示双侧额叶异常,伴有或不伴有顶叶的不同程度的变化;没有一个病例具有明显的局灶性或不对称性发现。14 名患者的神经病理学检查显示了一系列转激活反应 DNA 结合蛋白 43 型病理学(10 型 A 和 4 型 B),除了一个病例外,所有病例均有泛素阳性小脑颗粒神经元包涵体。9 名患者检测到运动神经元变性,包括 5 名无运动神经元病生前征象的患者。虽然存在变异性,但大多数携带该突变的患者具有特征性的人口统计学、临床、神经心理学、神经影像学和特别是神经病理学特征。