Bioinformatics Institute, A-STAR, Singapore.
BMC Genomics. 2011 Nov 30;12 Suppl 3(Suppl 3):S24. doi: 10.1186/1471-2164-12-S3-S24.
Lung cancer is the leading cause of cancer deaths in the world. The most common type of lung cancer is lung adenocarcinoma (AC). The genetic mechanisms of the early stages and lung AC progression steps are poorly understood. There is currently no clinically applicable gene test for the early diagnosis and AC aggressiveness. Among the major reasons for the lack of reliable diagnostic biomarkers are the extraordinary heterogeneity of the cancer cells, complex and poorly understudied interactions of the AC cells with adjacent tissue and immune system, gene variation across patient cohorts, measurement variability, small sample sizes and sub-optimal analytical methods. We suggest that gene expression profiling of the primary tumours and adjacent tissues (PT-AT) handled with a rational statistical and bioinformatics strategy of biomarker prediction and validation could provide significant progress in the identification of clinical biomarkers of AC. To minimise sample-to-sample variability, repeated multivariate measurements in the same object (organ or tissue, e.g. PT-AT in lung) across patients should be designed, but prediction and validation on the genome scale with small sample size is a great methodical challenge.
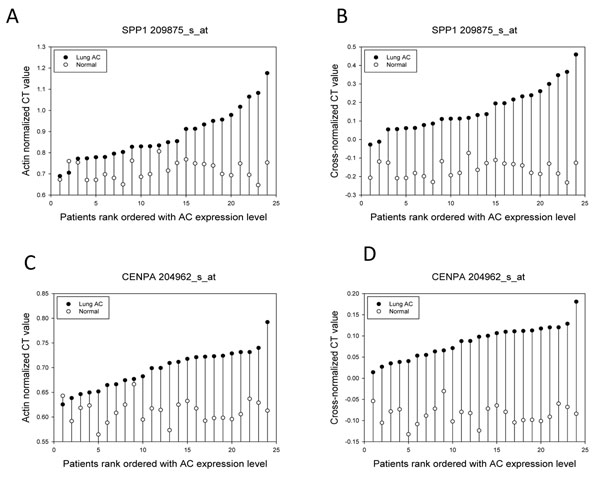
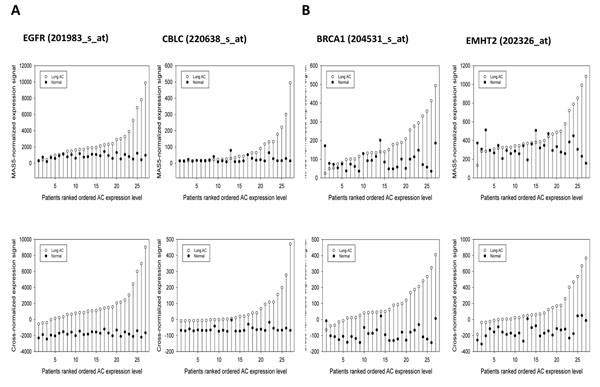
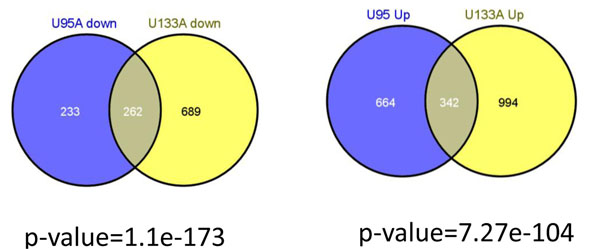
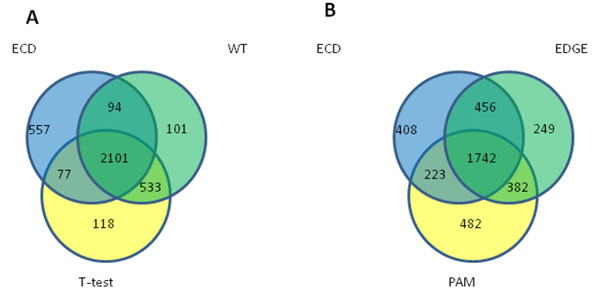
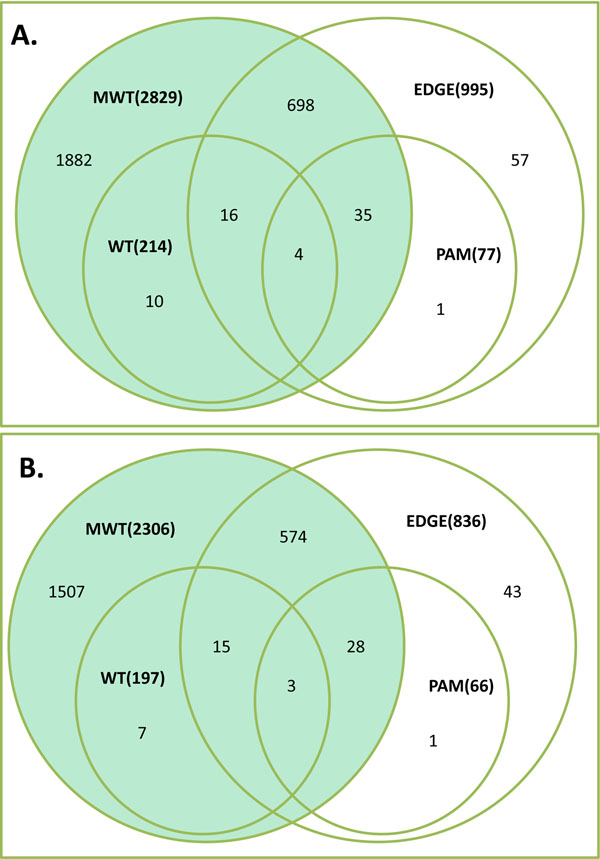
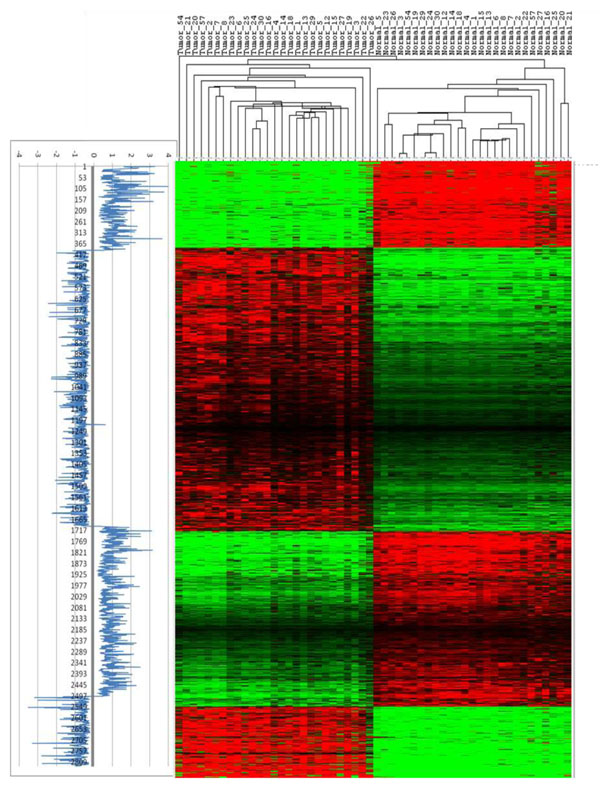
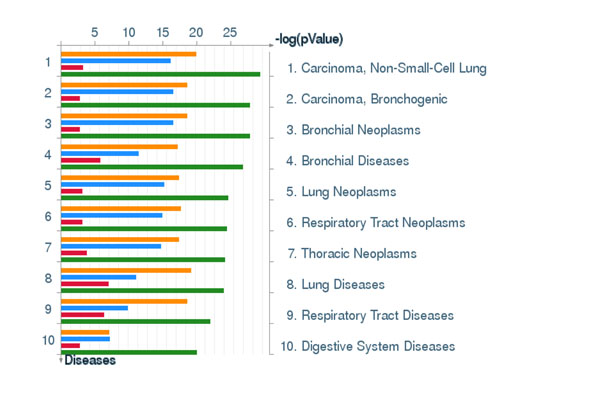
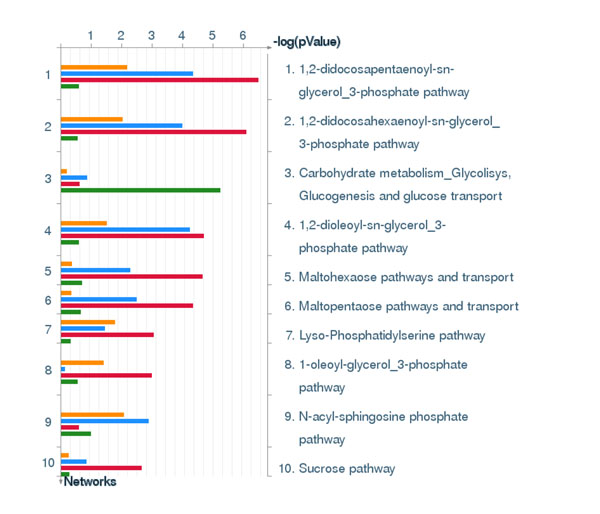
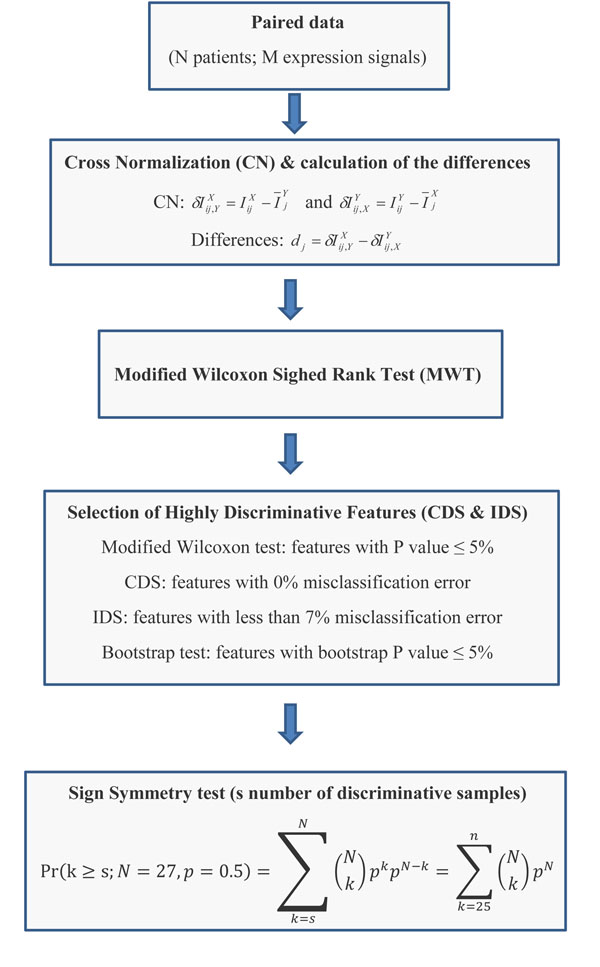
To analyse PT-AT relationships efficiently in the statistical modelling, we propose an Extreme Class Discrimination (ECD) feature selection method that identifies a sub-set of the most discriminative variables (e.g. expressed genes). Our method consists of a paired Cross-normalization (CN) step followed by a modified sign Wilcoxon test with multivariate adjustment carried out for each variable. Using an Affymetrix U133A microarray paired dataset of 27 AC patients, we reviewed the global reprogramming of the transcriptome in human lung AC tissue versus normal lung tissue, which is associated with about 2,300 genes discriminating the tissues with 100% accuracy. Cluster analysis applied to these genes resulted in four distinct gene groups which we classified as associated with (i) up-regulated genes in the mitotic cell cycle lung AC, (ii) silenced/suppressed gene specific for normal lung tissue, (iii) cell communication and cell motility and (iv) the immune system features. The genes related to mutagenesis, specific lung cancers, early stage of AC development, tumour aggressiveness and metabolic pathway alterations and adaptations of cancer cells are strongly enriched in the AC PT-AT discriminative gene set. Two AC diagnostic biomarkers SPP1 and CENPA were successfully validated on RT-RCR tissue array. ECD method was systematically compared to several alternative methods and proved to be of better performance and as well as it was validated by comparison of the predicted gene set with literature meta-signature.
We developed a method that identifies and selects highly discriminative variables from high dimensional data spaces of potential biomarkers based on a statistical analysis of paired samples when the number of samples is small. This method provides superior selection in comparison to conventional methods and can be widely used in different applications. Our method revealed at least 23 hundreds patho-biologically essential genes associated with the global transcriptional reprogramming of human lung epithelium cells and lung AC aggressiveness. This gene set includes many previously published AC biomarkers reflecting inherent disease complexity and specifies the mechanisms of carcinogenesis in the lung AC. SPP1, CENPA and many other PT-AT discriminative genes could be considered as the prospective diagnostic and prognostic biomarkers of lung AC.
肺癌是世界上癌症死亡的主要原因。最常见的肺癌类型是肺腺癌(AC)。肺癌早期阶段和 AC 进展步骤的遗传机制还了解甚少。目前,没有用于早期诊断和 AC 侵袭性的临床适用的基因检测。缺乏可靠诊断生物标志物的主要原因之一是癌细胞的非凡异质性、AC 细胞与相邻组织和免疫系统之间复杂且研究不足的相互作用、患者队列之间的基因变异、测量变异性、样本量小以及分析方法不理想。我们建议,通过对原发性肿瘤和相邻组织(PT-AT)进行基因表达谱分析,并采用合理的统计和生物信息学策略进行生物标志物预测和验证,可能会在识别 AC 的临床生物标志物方面取得重大进展。为了最大程度地减少样本间的变异性,应针对患者的同一对象(器官或组织,例如肺中的 PT-AT)设计重复的多变量测量,但在基因组范围内进行小样本量的预测和验证是一项重大的方法学挑战。
为了在统计建模中有效地分析 PT-AT 关系,我们提出了一种极端类别判别(ECD)特征选择方法,该方法可以识别出最具判别力的变量子集(例如表达基因)。我们的方法包括配对的交叉归一化(CN)步骤,然后对每个变量进行多元调整的改进符号 Wilcoxon 检验。使用 27 例 AC 患者的 Affymetrix U133A 微阵列配对数据集,我们回顾了人类肺 AC 组织与正常肺组织中转录组的全局重编程,该重编程与约 2300 个基因有关,这些基因可以 100%准确地区分组织。对这些基因进行聚类分析得到了四个不同的基因群,我们将其分类为与(i)肺 AC 有丝分裂细胞周期中上调的基因、(ii)正常肺组织中沉默/抑制的基因、(iii)细胞通讯和细胞运动以及(iv)免疫系统特征相关的基因。与致突变、特定肺癌、AC 早期发展、肿瘤侵袭性和代谢途径改变以及癌细胞适应相关的基因在 AC-PT-AT 判别基因集中强烈富集。两种 AC 诊断生物标志物 SPP1 和 CENPA 已成功通过 RT-RCR 组织阵列进行验证。ECD 方法与几种替代方法进行了系统比较,结果证明其性能更好,并且通过将预测基因集与文献荟萃签名进行比较进行了验证。
我们开发了一种方法,该方法基于对小样本量的配对样本的统计分析,从潜在生物标志物的高维数据空间中识别和选择高度判别性变量。与传统方法相比,该方法具有更好的选择性能,并且可以广泛应用于不同的应用。我们的方法揭示了至少 2300 个与人类肺上皮细胞和肺 AC 侵袭性的全局转录重编程相关的病理生物学必需基因。该基因集包括许多先前发表的 AC 生物标志物,反映了疾病的固有复杂性,并指定了肺 AC 中的致癌机制。SPP1、CENPA 和许多其他 PT-AT 判别基因可被视为肺 AC 的潜在诊断和预后生物标志物。