Maulik Ujjwal, Mallik Saurav, Mukhopadhyay Anirban, Bandyopadhyay Sanghamitra
Department of Computer Science and Engineering, Jadavpur University, Kolkata, West Bengal, India.
Machine Intelligence Unit, Indian Statistical Institute, Kolkata, West Bengal, India.
PLoS One. 2015 Apr 1;10(4):e0119448. doi: 10.1371/journal.pone.0119448. eCollection 2015.
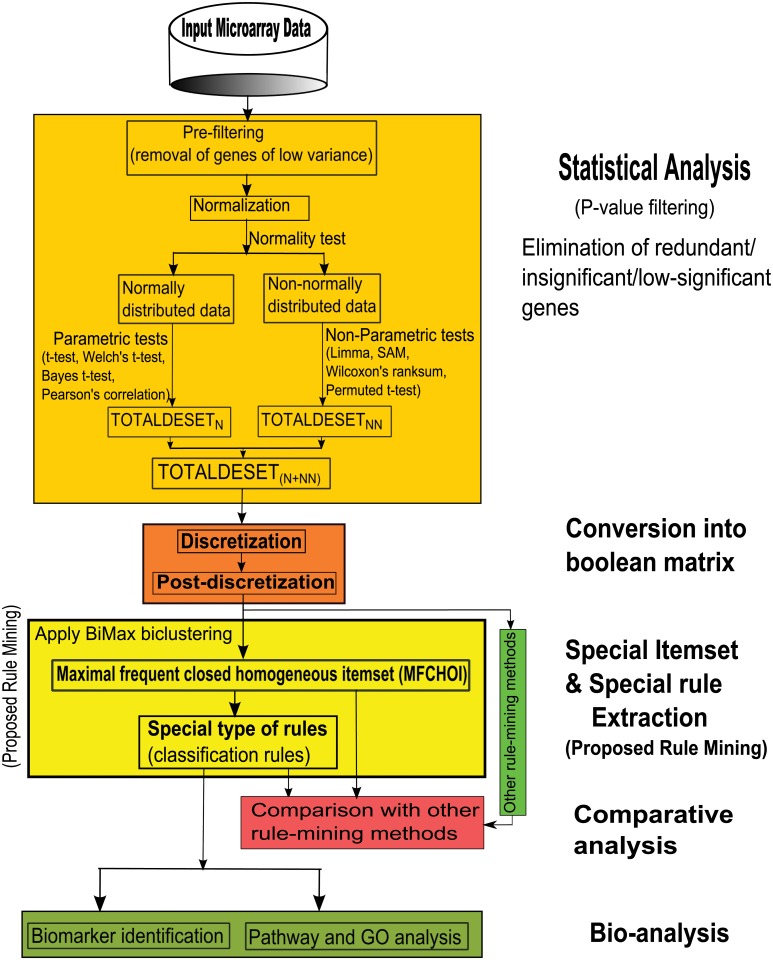
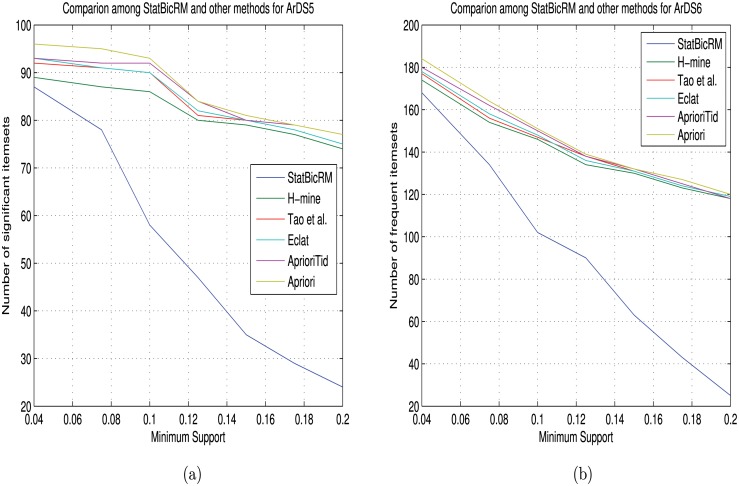
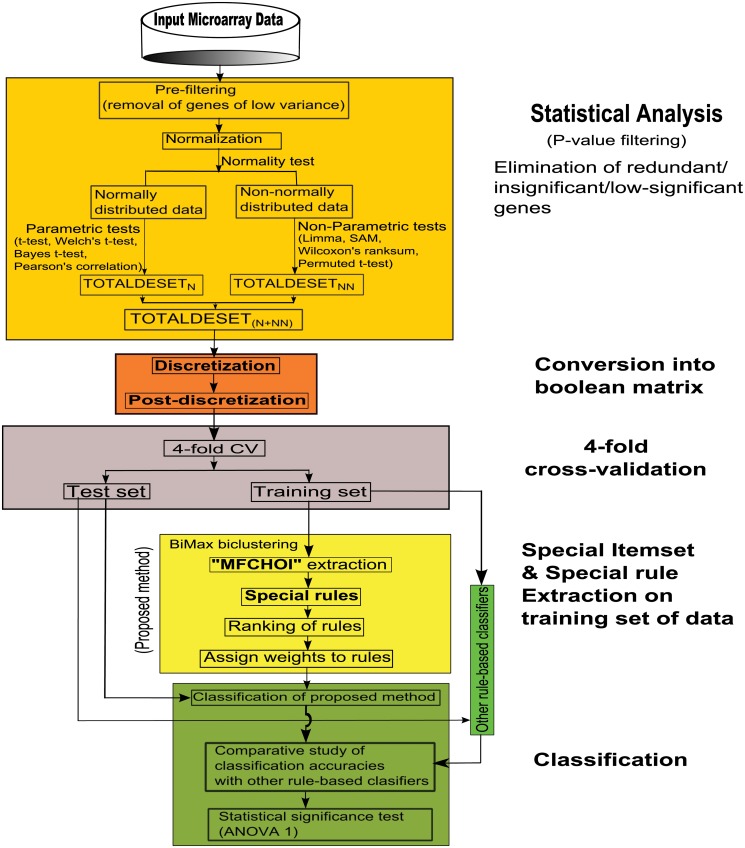
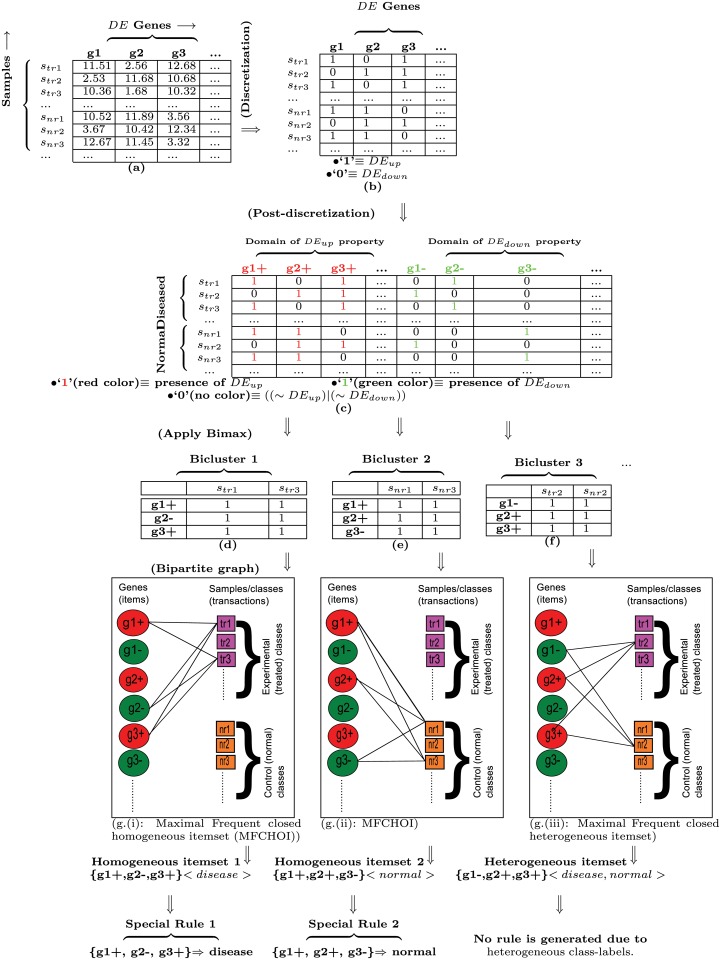
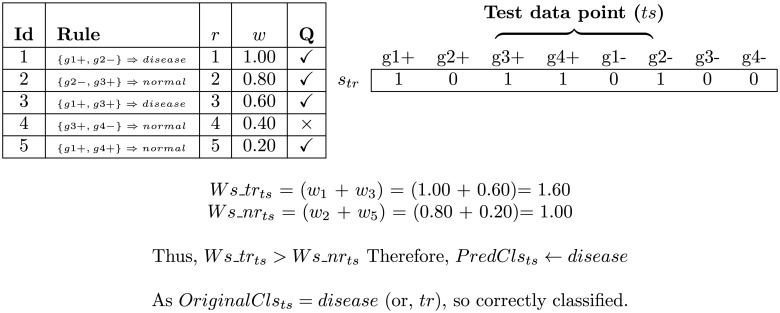
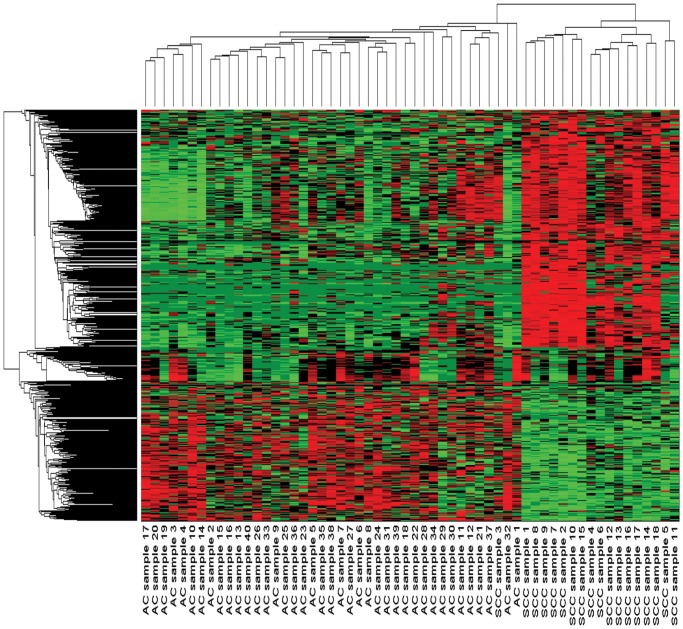
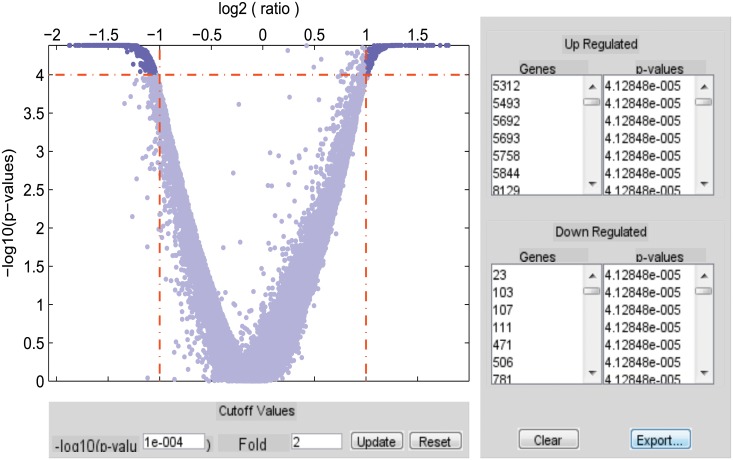
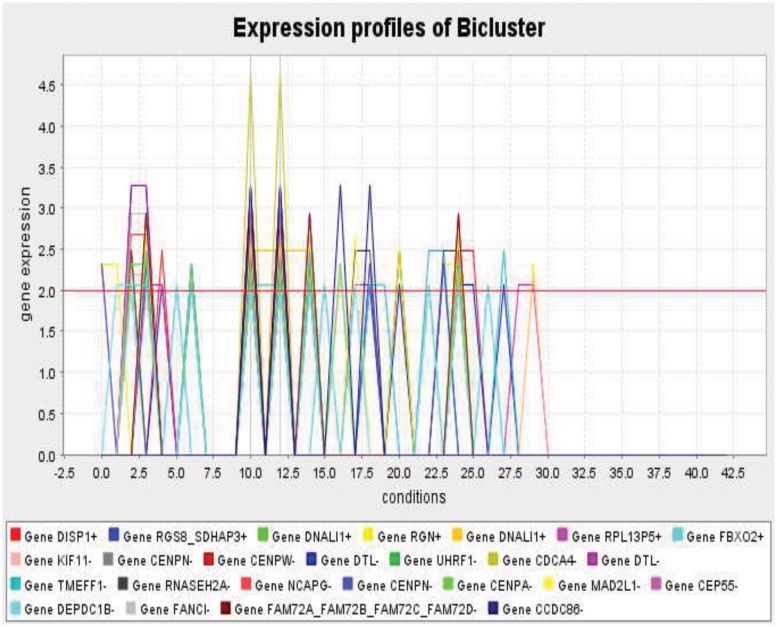
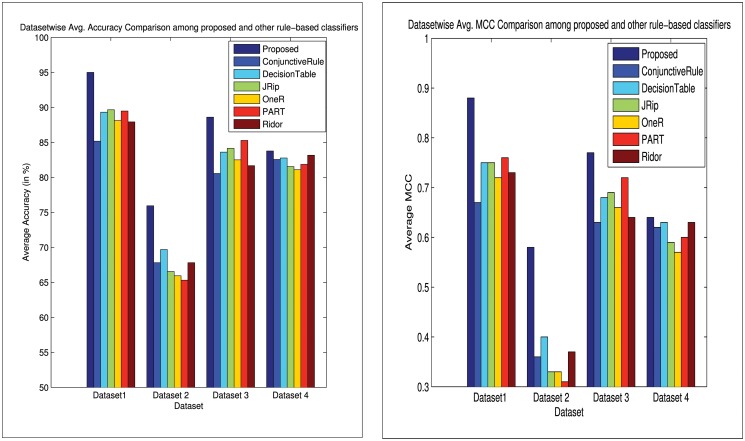
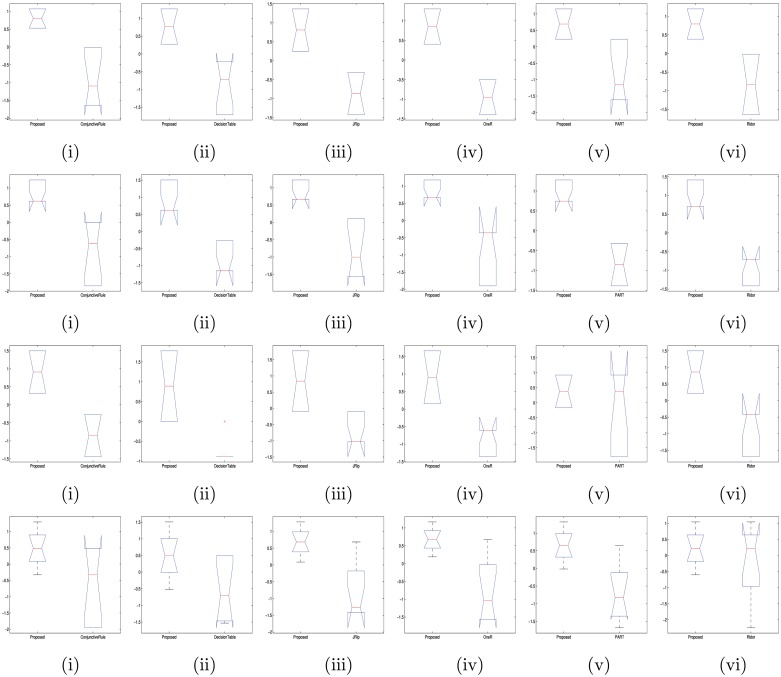
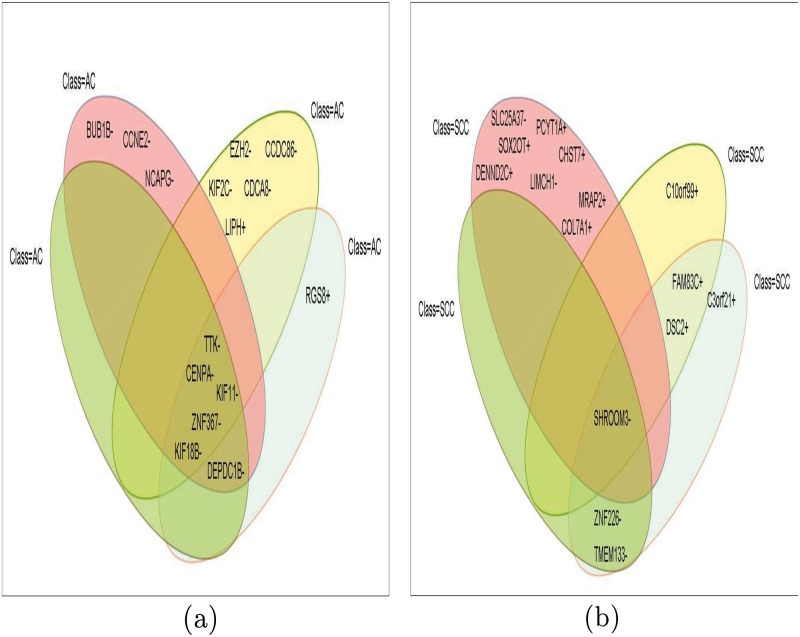
Microarray and beadchip are two most efficient techniques for measuring gene expression and methylation data in bioinformatics. Biclustering deals with the simultaneous clustering of genes and samples. In this article, we propose a computational rule mining framework, StatBicRM (i.e., statistical biclustering-based rule mining) to identify special type of rules and potential biomarkers using integrated approaches of statistical and binary inclusion-maximal biclustering techniques from the biological datasets. At first, a novel statistical strategy has been utilized to eliminate the insignificant/low-significant/redundant genes in such way that significance level must satisfy the data distribution property (viz., either normal distribution or non-normal distribution). The data is then discretized and post-discretized, consecutively. Thereafter, the biclustering technique is applied to identify maximal frequent closed homogeneous itemsets. Corresponding special type of rules are then extracted from the selected itemsets. Our proposed rule mining method performs better than the other rule mining algorithms as it generates maximal frequent closed homogeneous itemsets instead of frequent itemsets. Thus, it saves elapsed time, and can work on big dataset. Pathway and Gene Ontology analyses are conducted on the genes of the evolved rules using David database. Frequency analysis of the genes appearing in the evolved rules is performed to determine potential biomarkers. Furthermore, we also classify the data to know how much the evolved rules are able to describe accurately the remaining test (unknown) data. Subsequently, we also compare the average classification accuracy, and other related factors with other rule-based classifiers. Statistical significance tests are also performed for verifying the statistical relevance of the comparative results. Here, each of the other rule mining methods or rule-based classifiers is also starting with the same post-discretized data-matrix. Finally, we have also included the integrated analysis of gene expression and methylation for determining epigenetic effect (viz., effect of methylation) on gene expression level.
微阵列和珠芯片是生物信息学中用于测量基因表达和甲基化数据的两种最有效的技术。双聚类处理基因和样本的同时聚类。在本文中,我们提出了一种计算规则挖掘框架StatBicRM(即基于统计双聚类的规则挖掘),以使用来自生物数据集的统计和二元包含-最大双聚类技术的集成方法来识别特殊类型的规则和潜在的生物标志物。首先,利用一种新颖的统计策略以某种方式消除无意义/低显著性/冗余基因,使得显著性水平必须满足数据分布特性(即,正态分布或非正态分布)。然后,数据被依次离散化和后离散化。此后,应用双聚类技术来识别最大频繁封闭同质子集。然后从选定的子集中提取相应的特殊类型的规则。我们提出的规则挖掘方法比其他规则挖掘算法表现更好,因为它生成最大频繁封闭同质子集而不是频繁子集。因此,它节省了运行时间,并且可以处理大数据集。使用David数据库对进化规则的基因进行通路和基因本体分析。对出现在进化规则中的基因进行频率分析以确定潜在的生物标志物。此外,我们还对数据进行分类,以了解进化规则能够多准确地描述其余测试(未知)数据。随后,我们还将平均分类准确率和其他相关因素与其他基于规则的分类器进行比较。还进行统计显著性检验以验证比较结果的统计相关性。这里,其他每种规则挖掘方法或基于规则的分类器也都从相同的后离散化数据矩阵开始。最后,我们还纳入了基因表达和甲基化的综合分析,以确定表观遗传效应(即甲基化效应)对基因表达水平的影响。