National University of Singapore, 13 Computing Drive, Singapore 117417.
BMC Bioinformatics. 2011;12 Suppl 13(Suppl 13):S15. doi: 10.1186/1471-2105-12-S13-S15. Epub 2011 Nov 30.
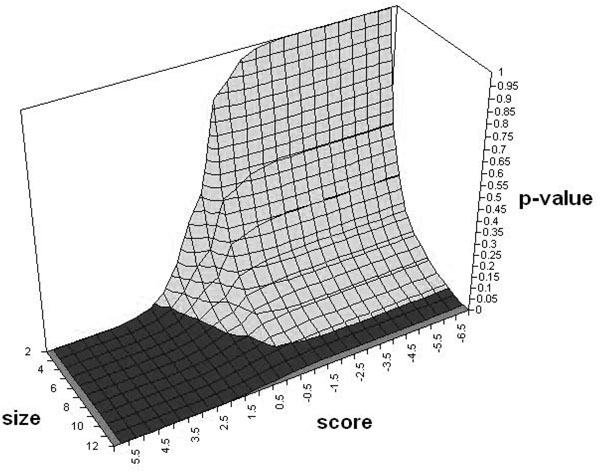
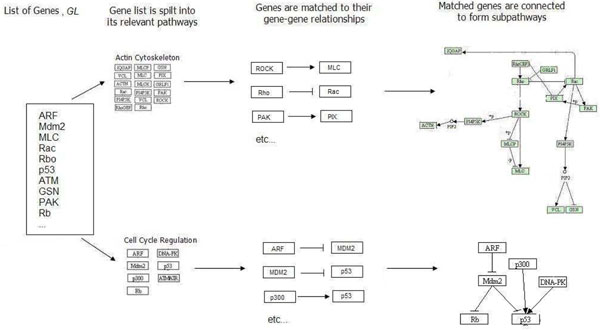
While contemporary methods of microarray analysis are excellent tools for studying individual microarray datasets, they have a tendency to produce different results from different datasets of the same disease. We aim to solve this reproducibility problem by introducing a technique (SNet). SNet provides both quantitative and descriptive analysis of microarray datasets by identifying specific connected portions of pathways that are significant. We term such portions within pathways as "subnetworks".
We tested SNet on independent datasets of several diseases, including childhood ALL, DMD and lung cancer. For each of these diseases, we obtained two independent microarray datasets produced by distinct labs on distinct platforms. In each case, our technique consistently produced almost the same list of significant nontrivial subnetworks from two independent sets of microarray data. The gene-level agreement of these significant subnetworks was between 51.18% to 93.01%. In contrast, when the same pairs of microarray datasets were analysed using GSEA, t-test and SAM, this percentage fell between 2.38% to 28.90% for GSEA, 49.60% tp 73.01% for t-test, and 49.96% to 81.25% for SAM. Furthermore, the genes selected using these existing methods did not form subnetworks of substantial size. Thus it is more probable that the subnetworks selected by our technique can provide the researcher with more descriptive information on the portions of the pathway actually affected by the disease.
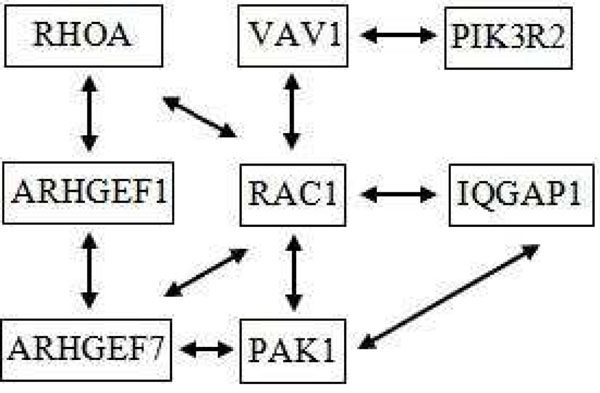
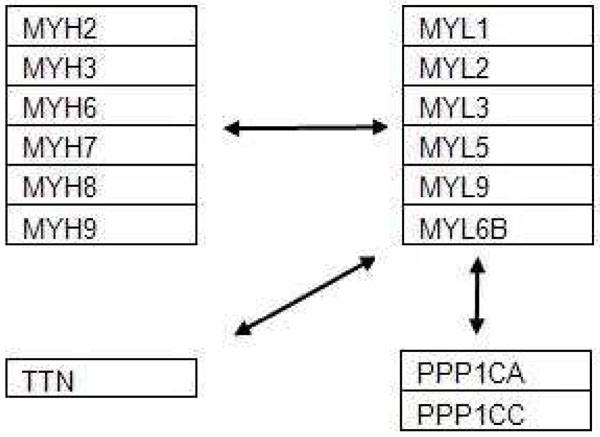
These results clearly demonstrate that our technique generates significant subnetworks and genes that are more consistent and reproducible across datasets compared to the other popular methods available (GSEA, t-test and SAM). The large size of subnetworks which we generate indicates that they are generally more biologically significant (less likely to be spurious). In addition, we have chosen two sample subnetworks and validated them with references from biological literature. This shows that our algorithm is capable of generating descriptive biologically conclusions.
虽然当代的微阵列分析方法是研究单个微阵列数据集的出色工具,但它们往往会从同一疾病的不同数据集产生不同的结果。我们旨在通过引入一种技术(SNet)来解决这个可重复性问题。SNet 通过识别有意义的特定连接路径部分,提供微阵列数据集的定量和描述性分析。我们将这些路径中的部分称为“子网”。
我们在包括儿童 ALL、DMD 和肺癌在内的几种疾病的独立数据集上测试了 SNet。对于每种疾病,我们都获得了两个由不同实验室在不同平台上产生的独立微阵列数据集。在每种情况下,我们的技术都从两组独立的微阵列数据中产生了几乎相同的显著非平凡子网列表。这些显著子网的基因水平一致性在 51.18%至 93.01%之间。相比之下,当使用 GSEA、t 检验和 SAM 分析相同的微阵列数据集对时,GSEA 的百分比在 2.38%至 28.90%之间,t 检验的百分比在 49.60%至 73.01%之间,SAM 的百分比在 49.96%至 81.25%之间。此外,使用这些现有方法选择的基因没有形成实质性大小的子网。因此,我们的技术选择的子网更有可能为研究人员提供有关疾病实际影响的通路部分的更具描述性的信息。
这些结果清楚地表明,与其他流行方法(GSEA、t 检验和 SAM)相比,我们的技术在数据集之间产生更一致和可重复的显著子网和基因。我们生成的子网的大规模表明它们通常更具有生物学意义(不太可能是虚假的)。此外,我们选择了两个示例子网,并通过生物学文献中的参考文献进行了验证。这表明我们的算法能够生成描述性的生物学结论。