G3 (Bethesda). 2011 Sep;1(4):303-16. doi: 10.1534/g3.111.000307. Epub 2011 Sep 1.
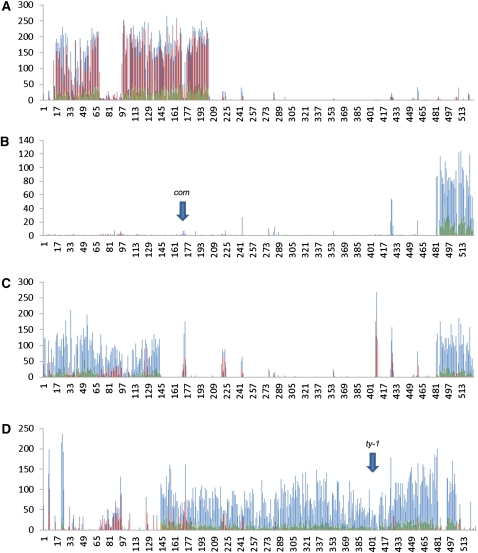
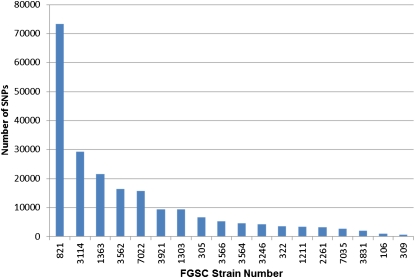
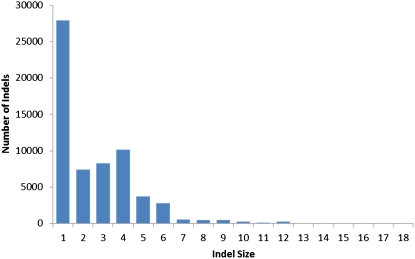
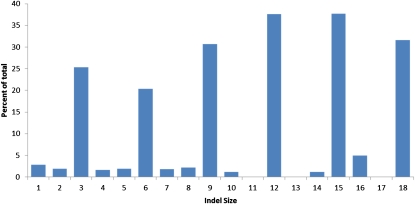
Classical forward genetics has been foundational to modern biology, and has been the paradigm for characterizing the role of genes in shaping phenotypes for decades. In recent years, reverse genetics has been used to identify the functions of genes, via the intentional introduction of variation and subsequent evaluation in physiological, molecular, and even population contexts. These approaches are complementary and whole genome analysis serves as a bridge between the two. We report in this article the whole genome sequencing of eighteen classical mutant strains of Neurospora crassa and the putative identification of the mutations associated with corresponding mutant phenotypes. Although some strains carry multiple unique nonsynonymous, nonsense, or frameshift mutations, the combined power of limiting the scope of the search based on genetic markers and of using a comparative analysis among the eighteen genomes provides strong support for the association between mutation and phenotype. For ten of the mutants, the mutant phenotype is recapitulated in classical or gene deletion mutants in Neurospora or other filamentous fungi. From thirteen to 137 nonsense mutations are present in each strain and indel sizes are shown to be highly skewed in gene coding sequence. Significant additional genetic variation was found in the eighteen mutant strains, and this variability defines multiple alleles of many genes. These alleles may be useful in further genetic and molecular analysis of known and yet-to-be-discovered functions and they invite new interpretations of molecular and genetic interactions in classical mutant strains.
经典正向遗传学是现代生物学的基础,几十年来一直是描述基因在塑造表型方面作用的典范。近年来,通过在生理、分子甚至种群背景下有意引入变异并随后进行评估,反向遗传学已被用于鉴定基因的功能。这些方法是互补的,全基因组分析则是两者之间的桥梁。我们在本文中报告了 18 株粗糙脉孢菌经典突变株的全基因组测序,并推测出与相应突变表型相关的突变。尽管一些菌株携带多个独特的非同义、无义或移码突变,但基于遗传标记限制搜索范围的综合能力,以及在 18 个基因组之间进行比较分析,为突变与表型之间的关联提供了强有力的支持。对于 10 种突变体,在粗糙脉孢菌或其他丝状真菌中的经典或基因缺失突变体中重现了突变体表型。每个菌株中存在 13 到 137 个无义突变,并且插入缺失大小在基因编码序列中显示出高度偏斜。在 18 个突变株中还发现了大量额外的遗传变异,这种可变性定义了许多基因的多个等位基因。这些等位基因可能对已知和尚未发现的功能的进一步遗传和分子分析有用,并为经典突变株中的分子和遗传相互作用提供了新的解释。