Craniofacial and Skeletal Diseases Branch, National Institute of Dental and Craniofacial Research, National Institutes of Health/DHHS, 9000 Rockville Pike, Bethesda, MD, USA 20892-4320, USA.
J Bone Miner Res. 2012 Jun;27(6):1309-21. doi: 10.1002/jbmr.1573.
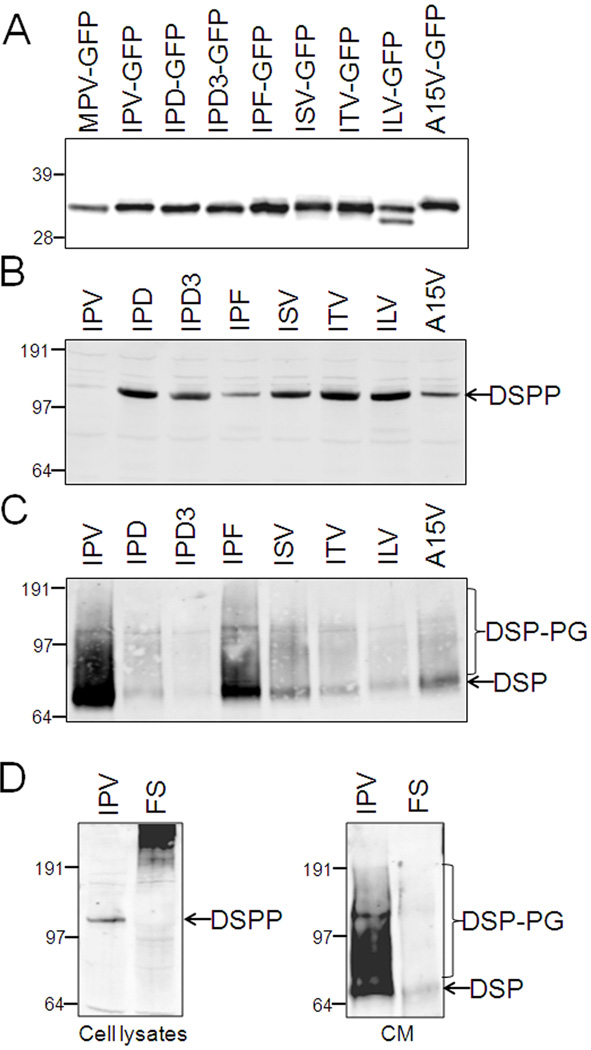
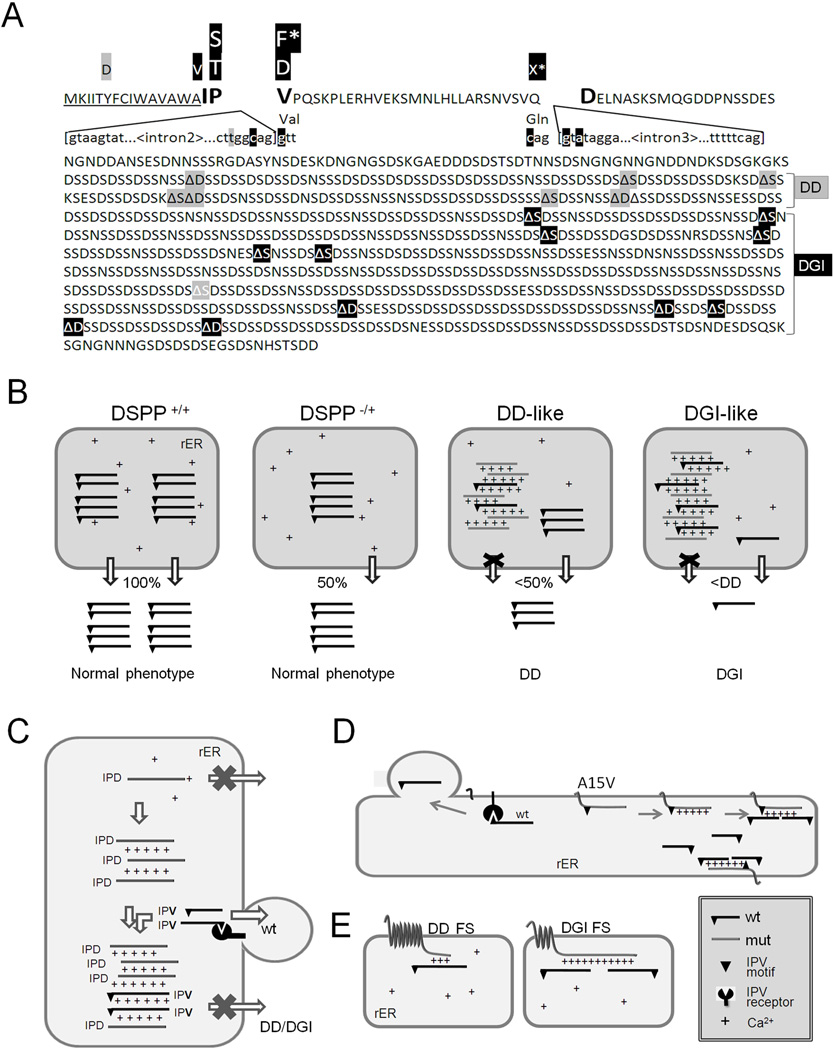
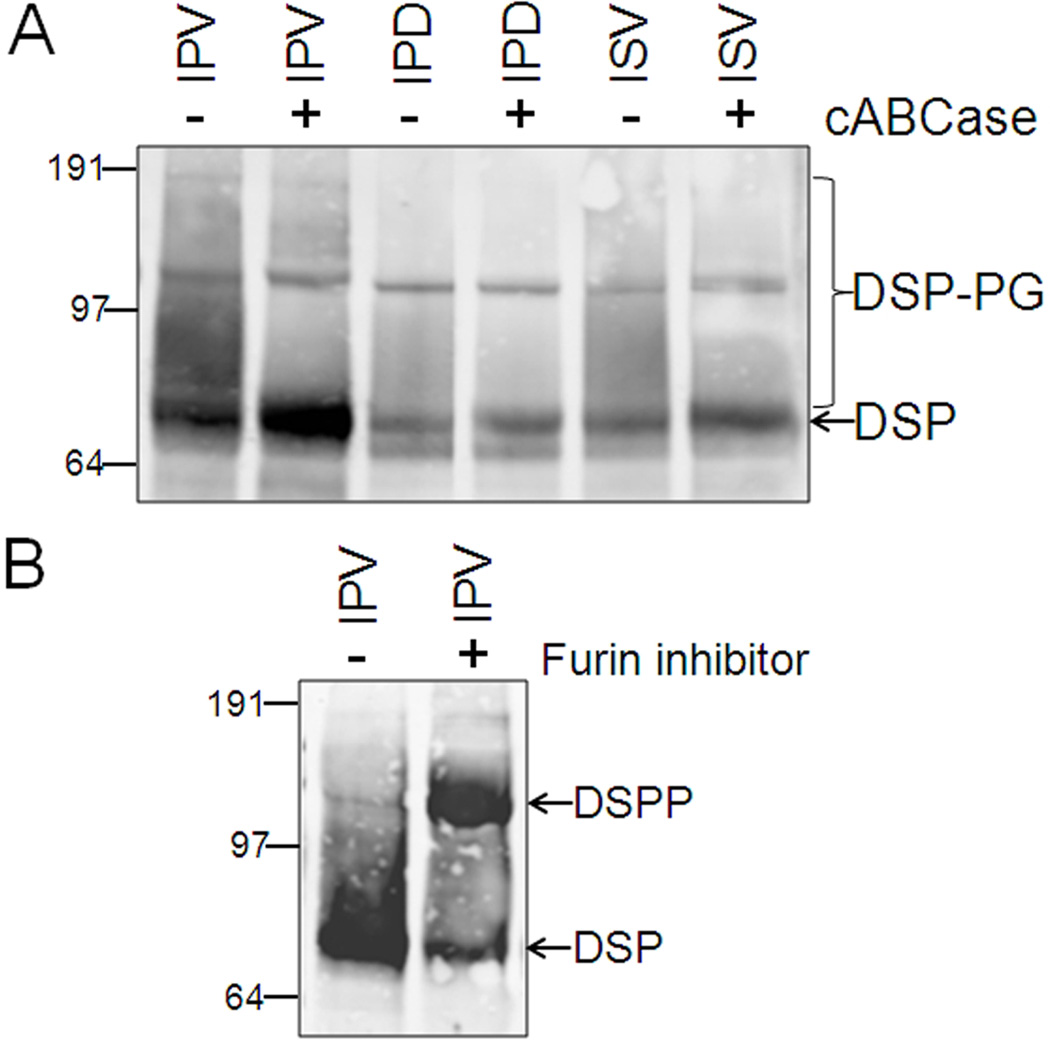
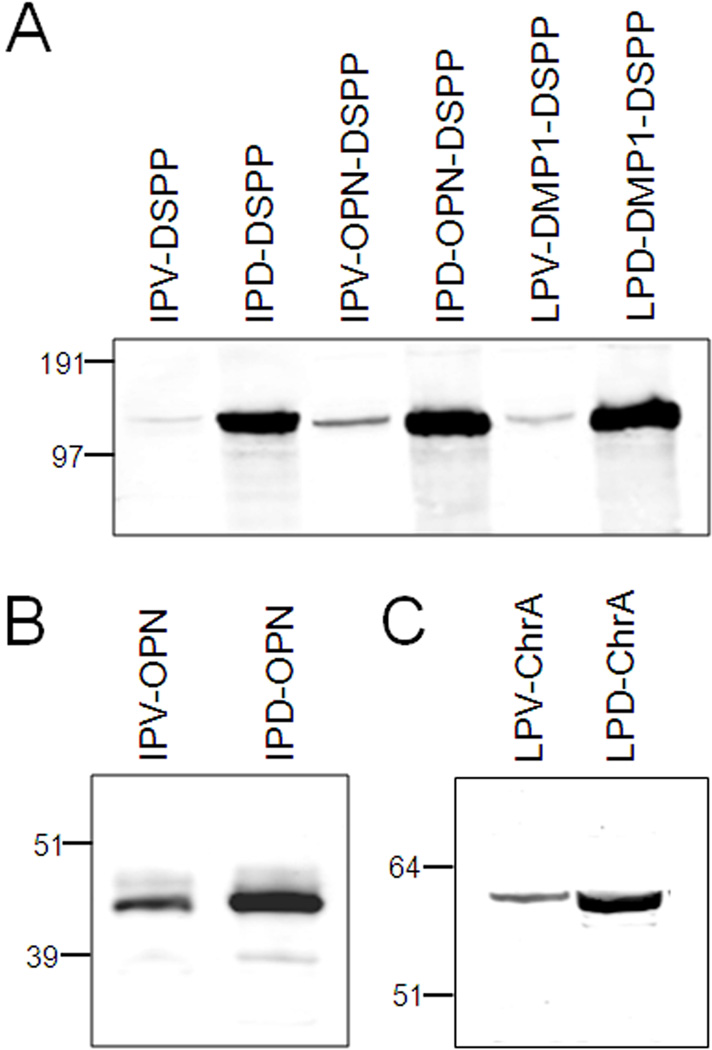
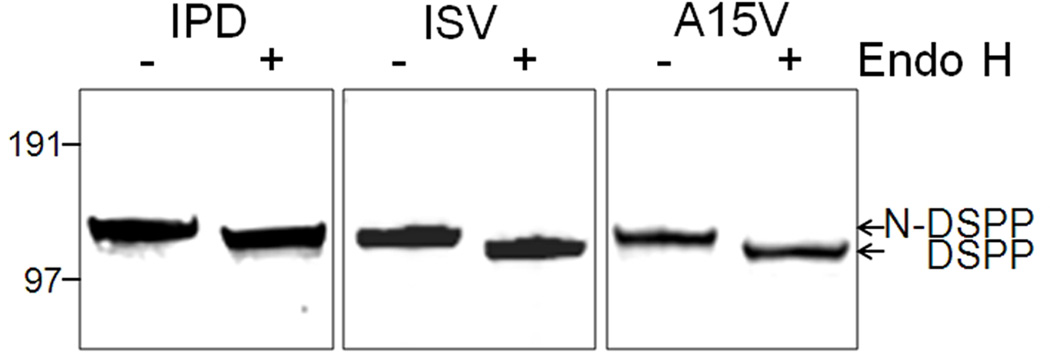
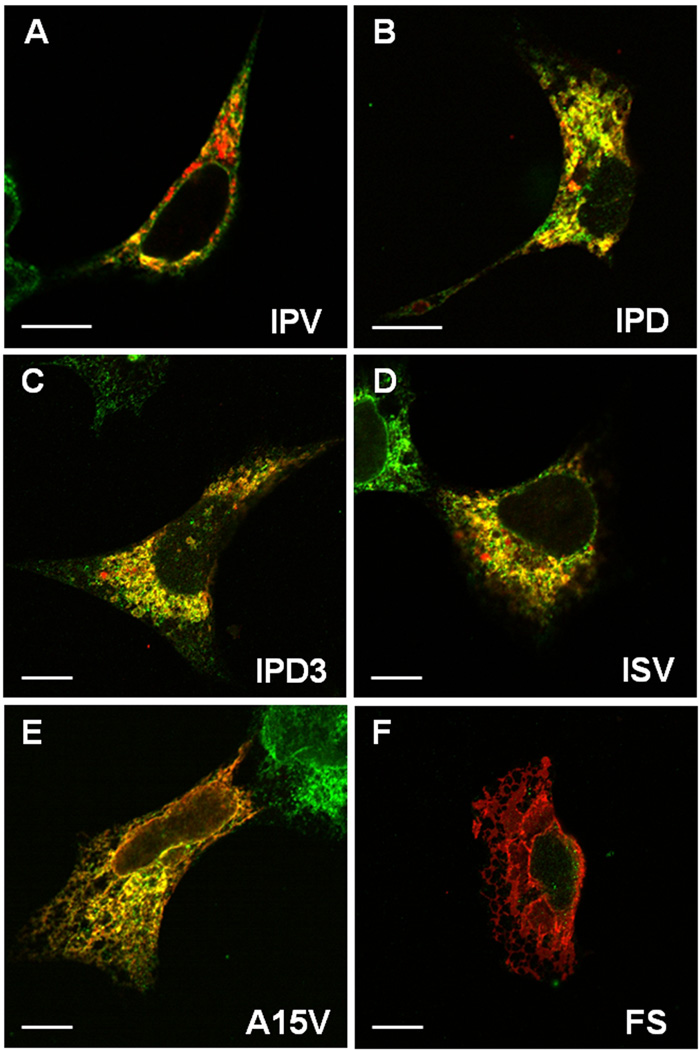
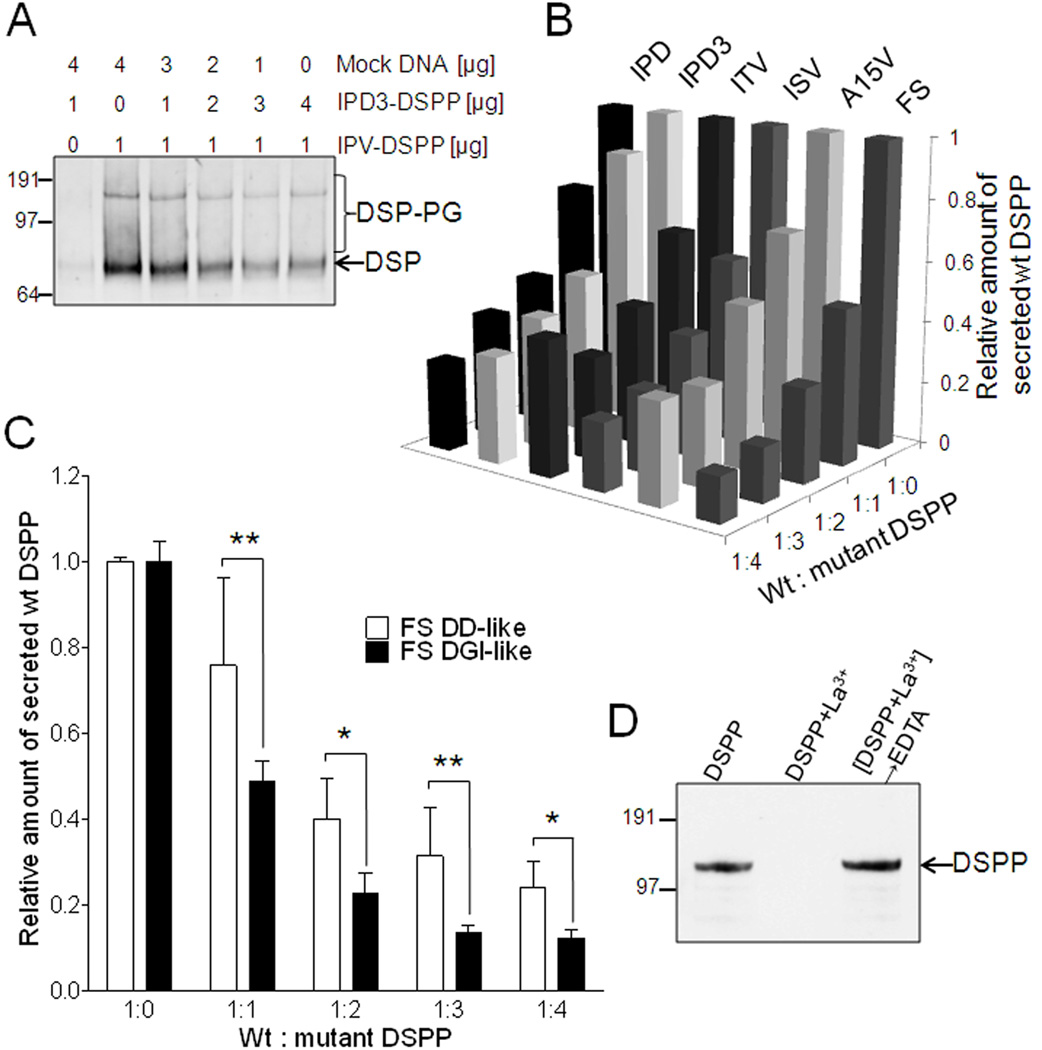
Families with nonsyndromic dentinogenesis imperfecta (DGI) and the milder, dentin dysplasia (DD), have mutations in one allele of the dentin sialophosphoprotein (DSPP) gene. Because loss of a single Dspp allele in mice (and likely, humans) causes no dental phenotype, the mechanism(s) underling the dominant negative effects were investigated. DSPP mutations occur in three classes. (The first class, the mid-leader missense mutation, Y6D, was not investigated in this report.) All other 5′ mutations of DSPP result in changes/loss in the first three amino acids (isoleucine-proline-valine [IPV]) of mature DSPP or, for the A15V missense mutation, some retention of the hydrophobic leader sequence. All of this second class of mutations caused mutant DSPP to be retained in the rough endoplasmic reticulum (rER) of transfected HEK293 cells. Trafficking out of the rER by coexpressed normal DSPP was reduced in a dose-responsive manner, probably due to formation of Ca2+-dependent complexes with the retained mutant DSPP. IPV-like sequences begin many secreted Ca2+-binding proteins, and changing the third amino acid to the charged aspartate (D) in three other acidic proteins also caused increased rER accumulation. Both the leader-retaining A15V and the long string of hydrophobic amino acids resulting from all known frameshift mutations within the 3′-encoded Ca2+-binding repeat domain (third class of mutations) caused retention by association of the mutant proteins with rER membranes. More 5′ frameshift mutations result in longer mutant hydrophobic domains, but the milder phenotype, DD, probably due to lower effectiveness of the remaining, shorter Ca2+-binding domain in capturing normal DSPP protein within the rER. This study presents evidence of a shared underlying mechanism of capturing of normal DSPP by two different classes of DSPP mutations and offers an explanation for the mild (DD-II) versus severe (DGI-II and III) nonsyndromic dentin phenotypes. Evidence is also presented that many acidic, Ca2+-binding proteins may use the same IPV-like receptor/pathway for exiting the rER.
非综合征型牙本质发育不全(DGI)和轻度牙本质发育不全(DD)的家族携带有 dentin sialophosphoprotein(DSPP)基因突变。由于在小鼠(可能还有人类)中缺失一个 Dspp 等位基因不会导致任何牙齿表型,因此研究了导致显性负效应的机制。DSPP 突变分为三类。(本报告未研究第一类中间先导区错义突变 Y6D。)DSPP 的所有其他 5′突变导致成熟 DSPP 的前三个氨基酸(异亮氨酸-脯氨酸-缬氨酸 [IPV])发生变化/缺失,或者对于 A15V 错义突变,保留一些疏水性先导序列。第二类所有突变导致突变型 DSPP 滞留在转染的 HEK293 细胞的粗面内质网(rER)中。通过共表达正常的 DSPP 从 rER 中的转运以剂量依赖性方式减少,这可能是由于与保留的突变型 DSPP 形成 Ca2+依赖性复合物所致。像 IPV 这样的序列开始许多分泌型 Ca2+结合蛋白,并且将三个其他酸性蛋白中的第三个氨基酸改变为带电荷的天冬氨酸(D)也会导致 rER 积累增加。A15V 保留先导区和所有已知的 3′编码 Ca2+结合重复结构域内的框架移位突变导致的长疏水区段(第三类突变)都导致突变蛋白与 rER 膜结合而滞留。更多的 5′框架移位突变导致更长的突变疏水区段,但表型较轻,DD,可能是由于较短的 Ca2+结合结构域在 rER 中捕获正常 DSPP 蛋白的效率较低所致。本研究提供了证据证明两种不同类型的 DSPP 突变通过共同的潜在机制捕获正常 DSPP,并为非综合征性牙本质表型的轻度(DD-II)与重度(DGI-II 和 III)提供了解释。还提出了证据表明,许多酸性、Ca2+结合蛋白可能使用相同的 IPV 样受体/途径从 rER 中输出。