Multidisciplinary Centre for Integrative Biology (MyCIB), School of Biosciences, University of Nottingham, Nottingham, UK.
Nucleic Acids Res. 2012 Jul;40(12):5227-39. doi: 10.1093/nar/gks205. Epub 2012 Mar 9.
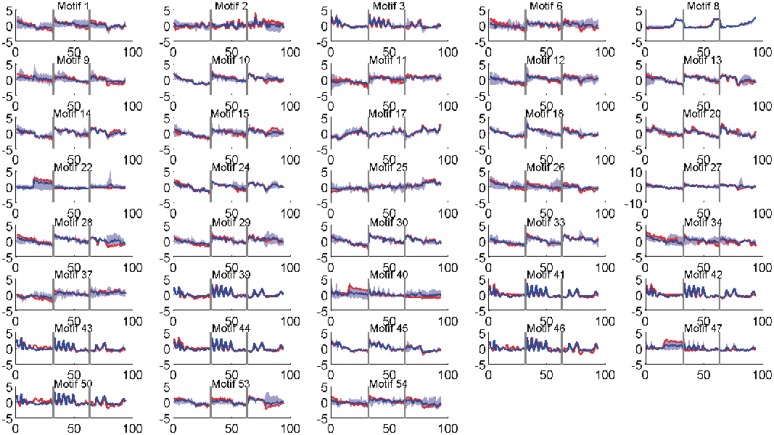
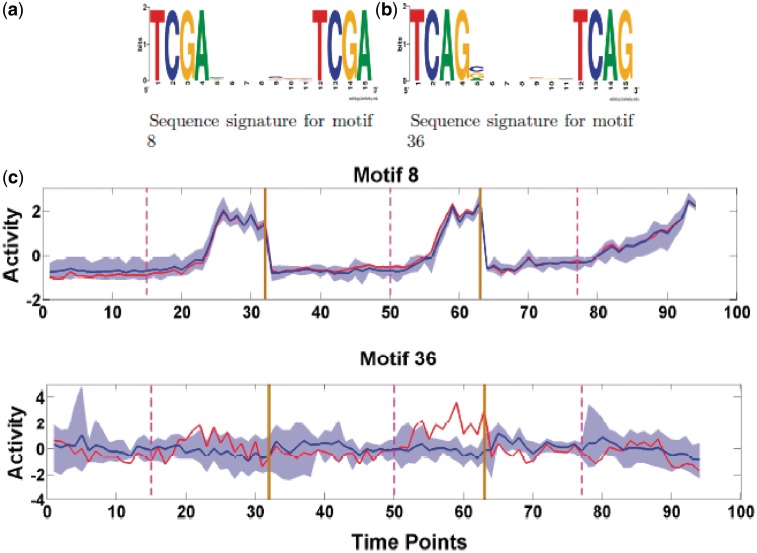
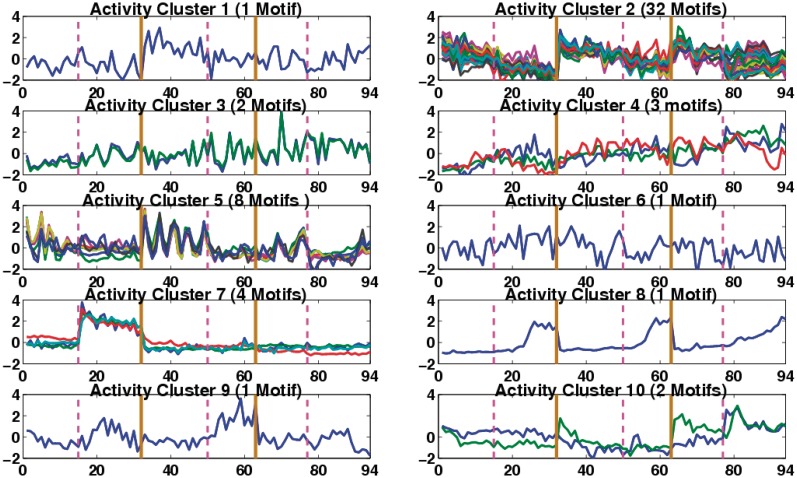
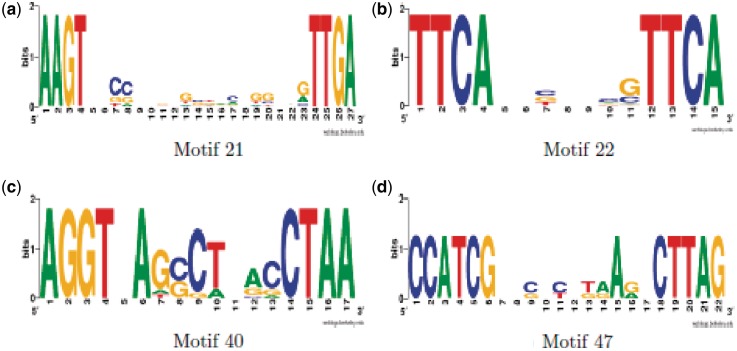
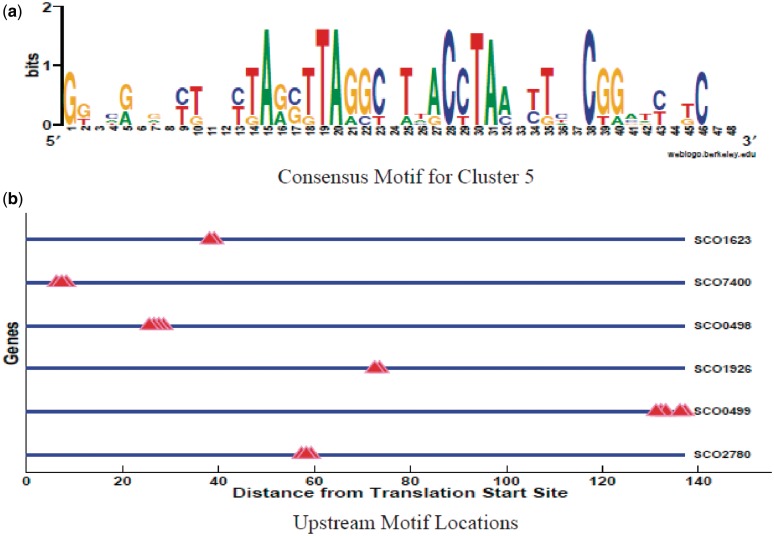
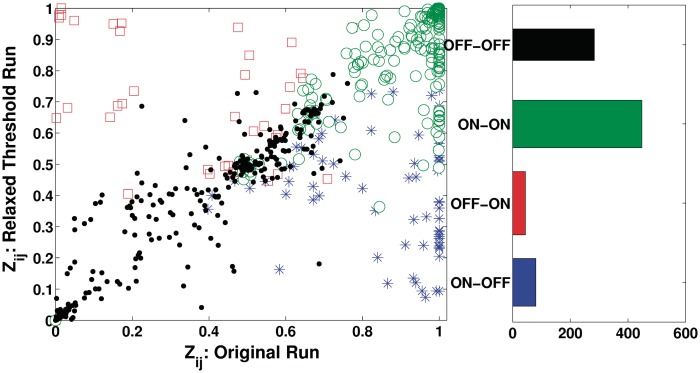
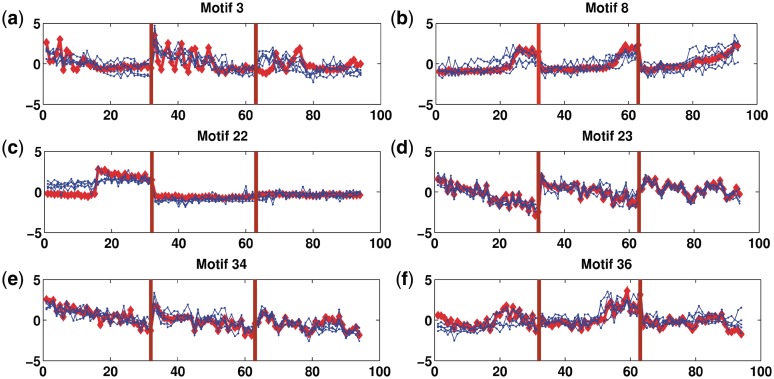
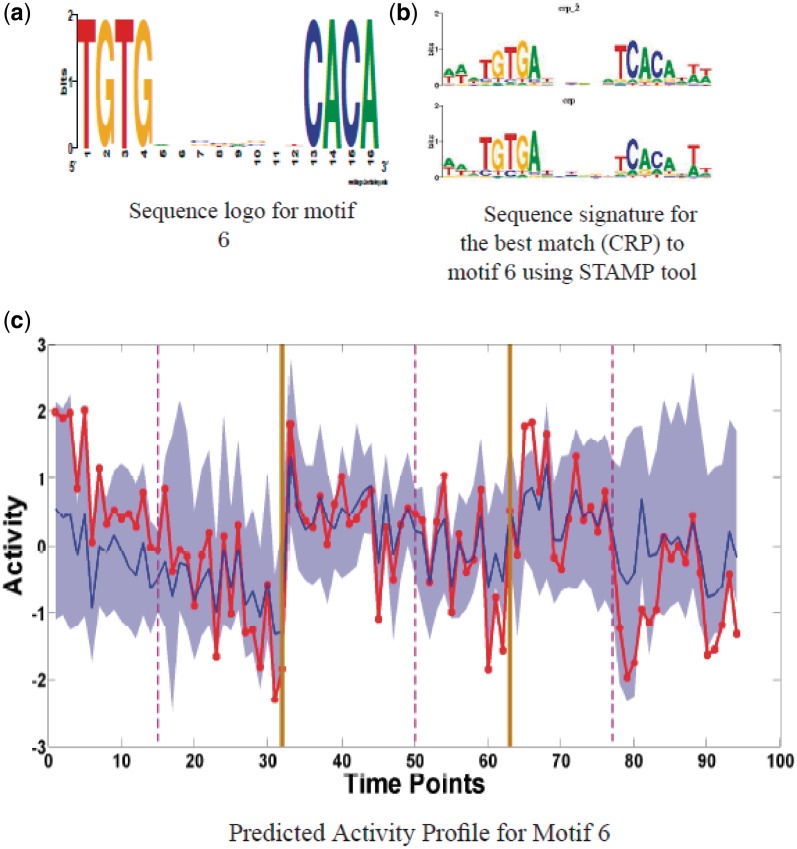
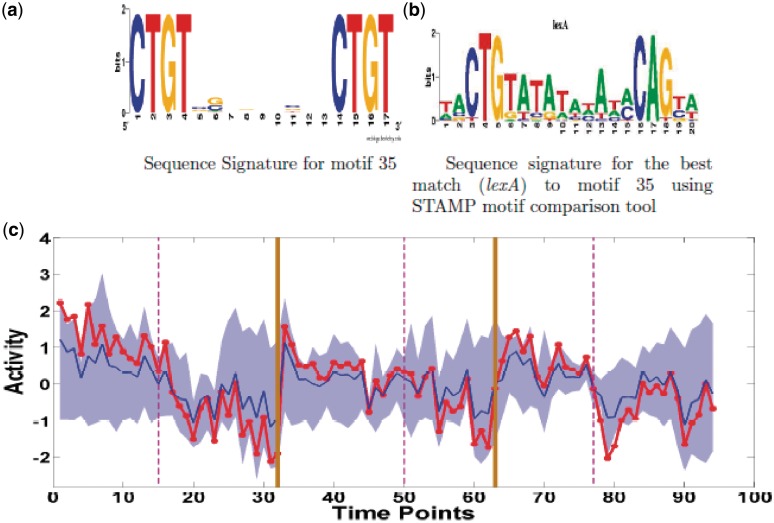
Determining transcriptional regulator activities is a major focus of systems biology, providing key insight into regulatory mechanisms and co-regulators. For organisms such as Escherichia coli, transcriptional regulator binding site data can be integrated with expression data to infer transcriptional regulator activities. However, for most organisms there is only sparse data on their transcriptional regulators, while their associated binding motifs are largely unknown. Here, we address the challenge of inferring activities of unknown regulators by generating de novo (binding) motifs and integrating with expression data. We identify a number of key regulators active in the metabolic switch, including PhoP with its associated directed repeat PHO box, candidate motifs for two SARPs, a CRP family regulator, an iron response regulator and that for LexA. Experimental validation for some of our predictions was obtained using gel-shift assays. Our analysis is applicable to any organism for which there is a reasonable amount of complementary expression data and for which motifs (either over represented or evolutionary conserved) can be identified in the genome.
确定转录调控因子的活性是系统生物学的主要关注点,为研究调控机制和共调控因子提供了关键的见解。对于像大肠杆菌这样的生物体,可以将转录调控因子结合位点的数据与表达数据进行整合,以推断转录调控因子的活性。然而,对于大多数生物体来说,关于它们的转录调控因子的数据很少,而它们相关的结合基序在很大程度上是未知的。在这里,我们通过生成新的(结合)基序并与表达数据进行整合来解决推断未知调控因子活性的挑战。我们确定了一些在代谢开关中起关键作用的调控因子,包括 PhoP 及其相关的定向重复 PHO 盒、两个 SARPs 的候选基序、CRP 家族调控因子、一个铁反应调控因子和 LexA 的基序。我们的一些预测的实验验证是通过凝胶迁移分析获得的。我们的分析适用于任何具有相当数量的互补表达数据的生物体,并且可以在基因组中识别出(过度表达或进化保守的)基序。