Tropical and Emerging Infectious Diseases Division, Menzies School of Health Research, Charles Darwin University, Darwin, Northern Territory, Australia.
PLoS One. 2012;7(3):e33530. doi: 10.1371/journal.pone.0033530. Epub 2012 Mar 12.
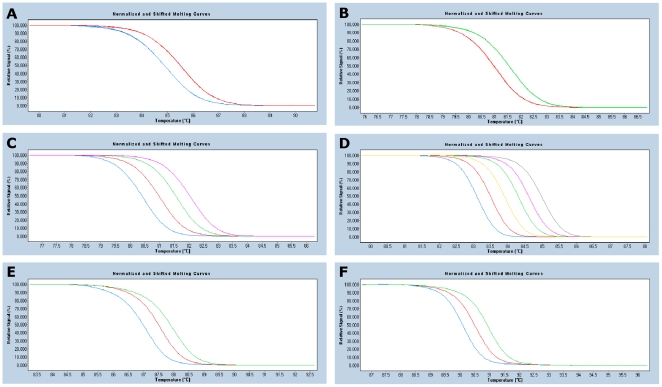
Here we report a single nucleotide polymorphism (SNP) based genotyping method for Klebsiella pneumoniae utilising high-resolution melting (HRM) analysis of fragments within the multilocus sequence typing (MLST) loci. The approach is termed mini-MLST or Minim typing and it has previously been applied to Streptococcus pyogenes, Staphylococcus aureus and Enterococcus faecium. Six SNPs were derived from concatenated MLST sequences on the basis of maximisation of the Simpsons Index of Diversity (D). DNA fragments incorporating these SNPs and predicted to be suitable for HRM analysis were designed. Using the assumption that HRM alleles are defined by G+C content, Minim typing using six fragments was predicted to provide a D = 0.979 against known STs. The method was tested against 202 K. pneumoniae using a blinded approach in which the MLST analyses were performed after the HRM analyses. The HRM-based alleles were indeed in accordance with G+C content, and the Minim typing identified known STs and flagged new STs. The tonB MLST locus was determined to be very diverse, and the two Minim fragments located herein contribute greatly to the resolving power. However these fragments are refractory to amplification in a minority of isolates. Therefore, we assessed the performance of two additional formats: one using only the four fragments located outside the tonB gene (D = 0.929), and the other using HRM data from these four fragments in conjunction with sequencing of the tonB MLST fragment (D = 0.995). The HRM assays were developed on the Rotorgene 6000, and the method was shown to also be robust on the LightCycler 480, allowing a 384-well high through-put format. The assay provides rapid, robust and low-cost typing with fully portable results that can directly be related to current MLST data. Minim typing in combination with molecular screening for antibiotic resistance markers can be a powerful surveillance tool kit.
我们在此报告了一种基于单核苷酸多态性(SNP)的肺炎克雷伯菌基因分型方法,利用多位点序列分型(MLST)基因座内片段的高分辨率熔解(HRM)分析。该方法称为 mini-MLST 或 Minim 分型,先前已应用于化脓性链球菌、金黄色葡萄球菌和屎肠球菌。根据 Simpson 多样性指数(D)的最大化,从串联 MLST 序列中得出了 6 个 SNP。设计了包含这些 SNP 且预测适合 HRM 分析的 DNA 片段。假设 HRM 等位基因由 G+C 含量定义,使用 6 个片段的 Minim 分型预计提供 D=0.979 以对抗已知 ST。该方法使用 202 株肺炎克雷伯菌进行了测试,采用 HRM 分析后进行 MLST 分析的盲法。基于 HRM 的等位基因确实与 G+C 含量一致,Minim 分型识别了已知 ST 并标记了新的 ST。tonB MLST 基因座非常多样化,位于此处的两个 Minim 片段对分辨率有很大贡献。然而,这些片段在少数分离株中难以扩增。因此,我们评估了两种其他格式的性能:一种仅使用位于 tonB 基因之外的四个片段(D=0.929),另一种使用这四个片段的 HRM 数据结合 tonB MLST 片段的测序(D=0.995)。HRM 测定在 Rotorgene 6000 上进行,并且该方法在 LightCycler 480 上也显示出稳健性,允许 384 孔高通量格式。该测定提供了快速、稳健和低成本的分型,具有完全便携的结果,可以直接与当前的 MLST 数据相关联。Minim 分型与抗生素耐药性标记物的分子筛选相结合,可以成为一种强大的监测工具包。