INSERM U974, Institut de Myologie, Paris, France.
J Mol Cell Cardiol. 2012 Jun;52(6):1299-307. doi: 10.1016/j.yjmcc.2012.03.009. Epub 2012 Mar 23.
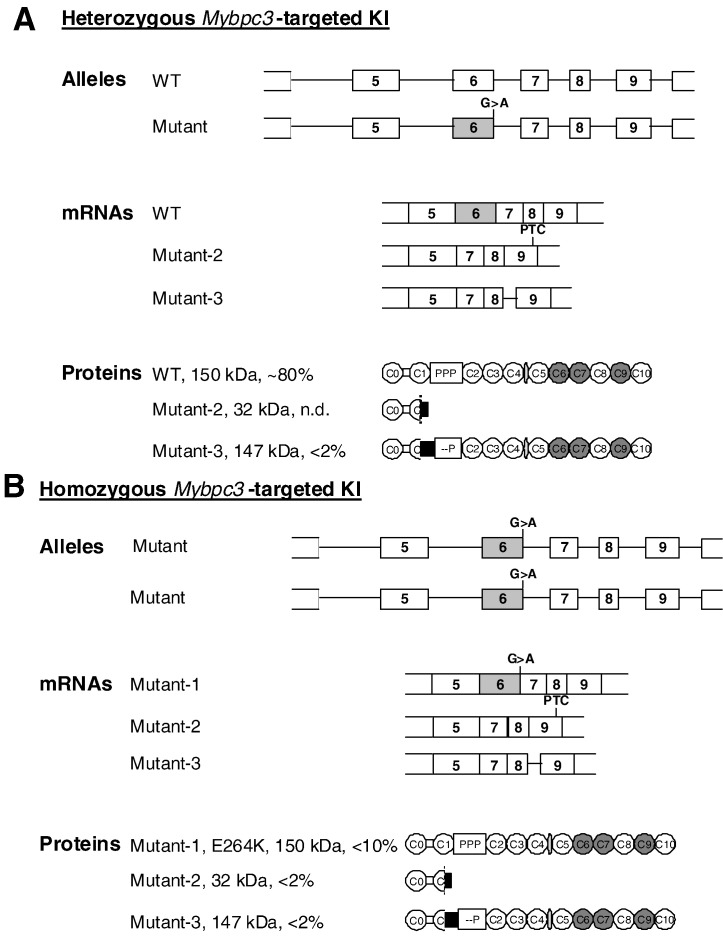
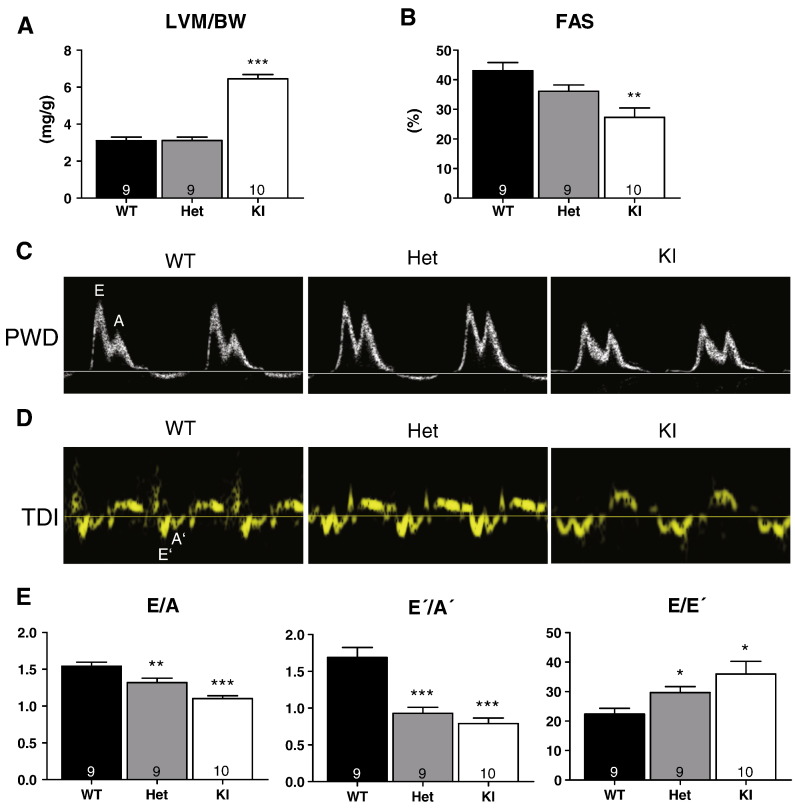
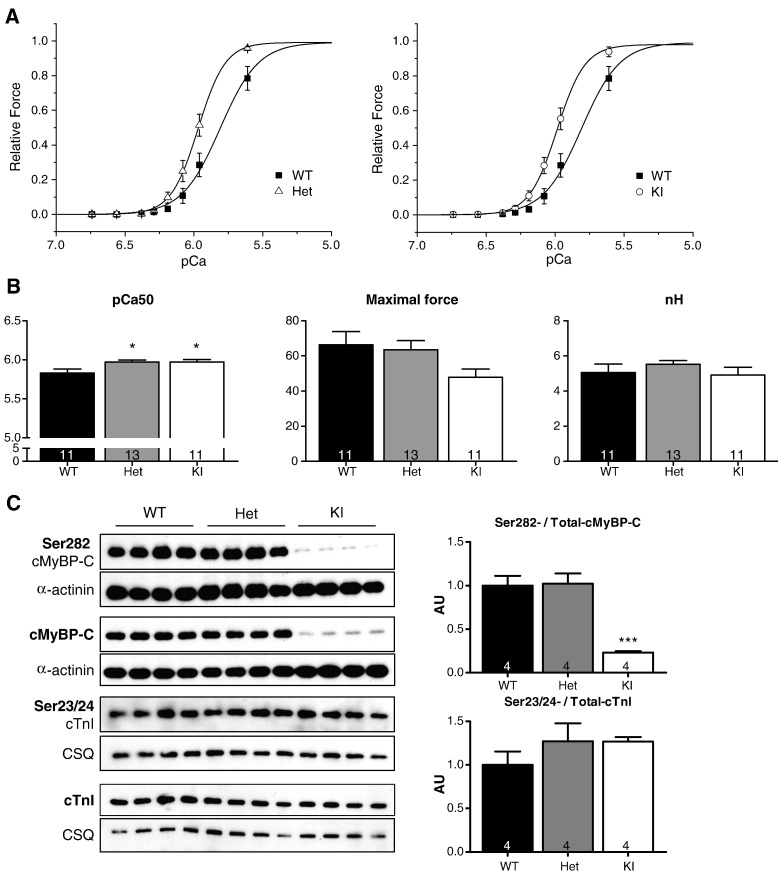
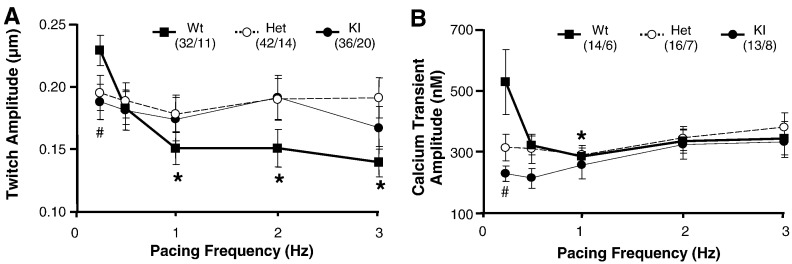
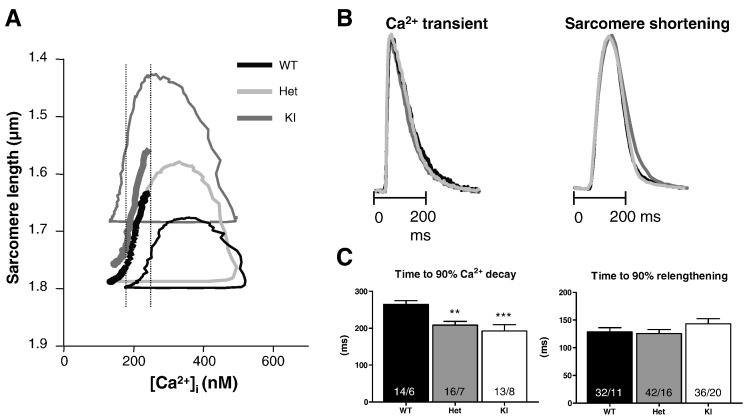
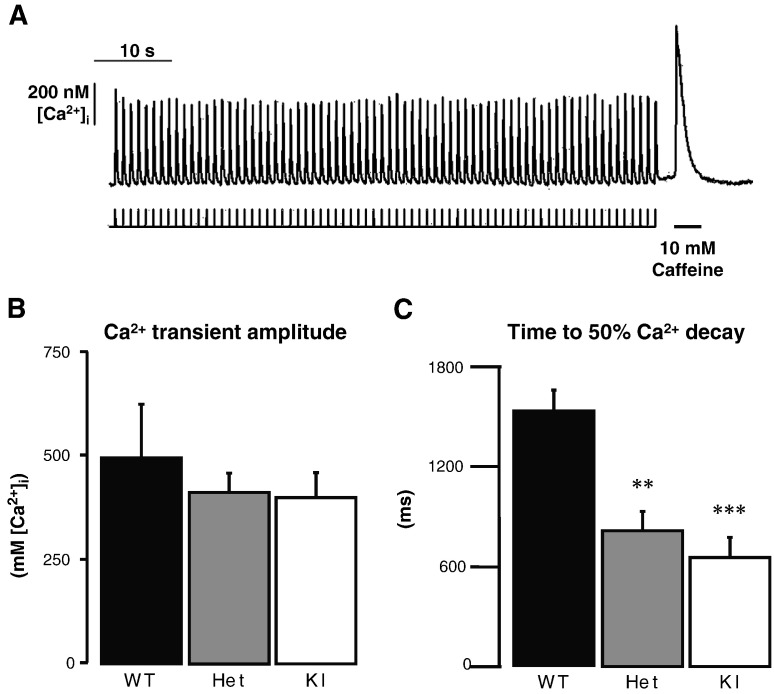
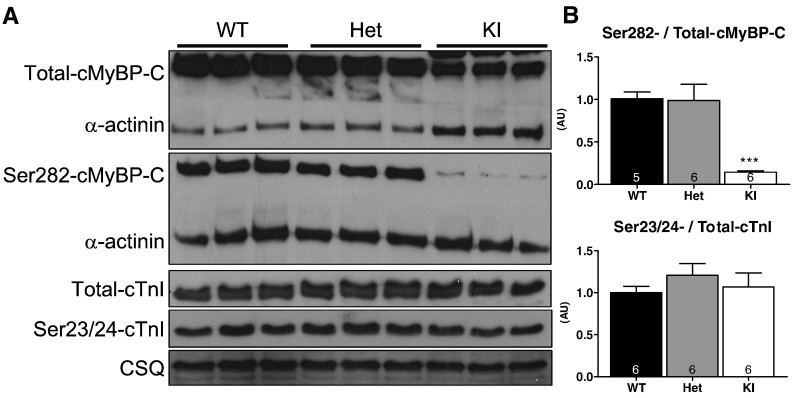
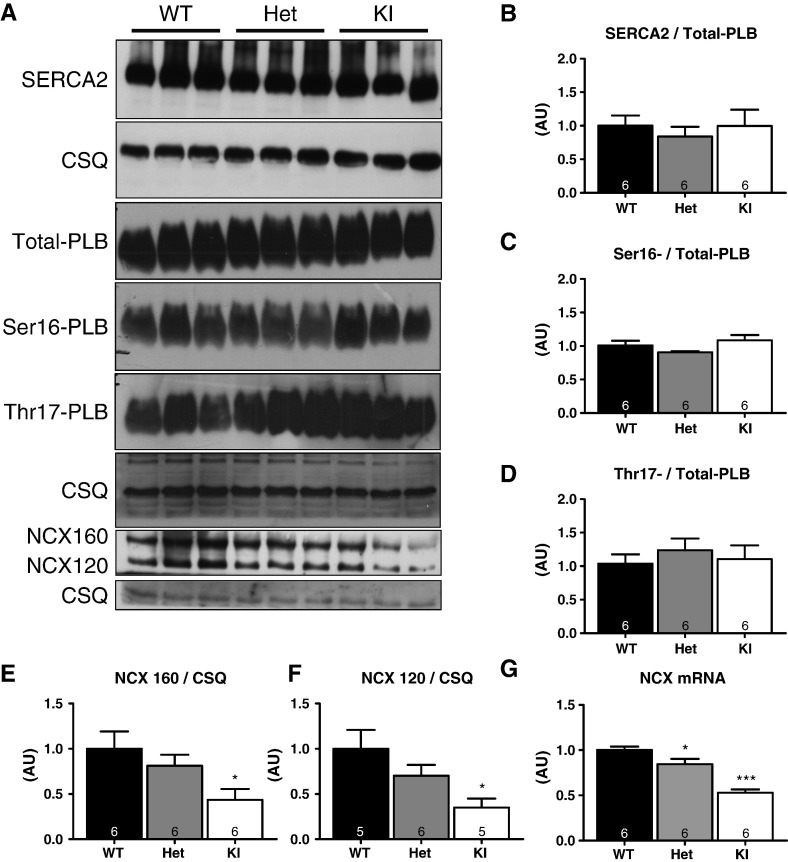
Hypertrophic cardiomyopathy (HCM) is frequently caused by mutations in MYBPC3 encoding cardiac myosin-binding protein C (cMyBP-C). The mechanisms leading from gene mutations to the HCM phenotype remain incompletely understood, partially because current mouse models of HCM do not faithfully reflect the human situation and early hypertrophy confounds the interpretation of functional alterations. The goal of this study was to evaluate whether myofilament Ca(2+) sensitization and diastolic dysfunction are associated or precede the development of left ventricular hypertrophy (LVH) in HCM. We evaluated the function of skinned and intact cardiac myocytes, as well as the intact heart in a recently developed Mybpc3-targeted knock-in mouse model carrying a point mutation frequently associated with HCM. Compared to wild-type, 10-week old homozygous knock-in mice exhibited i) higher myofilament Ca(2+) sensitivity in skinned ventricular trabeculae, ii) lower diastolic sarcomere length, and faster Ca(2+) transient decay in intact myocytes, and iii) LVH, reduced fractional shortening, lower E/A and E'/A', and higher E/E' ratios by echocardiography and Doppler analysis, suggesting systolic and diastolic dysfunction. In contrast, heterozygous knock-in mice, which mimic the human HCM situation, did not exhibit LVH or systolic dysfunction, but exhibited higher myofilament Ca(2+) sensitivity, faster Ca(2+) transient decay, and diastolic dysfunction. These data demonstrate that myofilament Ca(2+) sensitization and diastolic dysfunction are early phenotypic consequences of Mybpc3 mutations independent of LVH. The accelerated Ca(2+) transients point to compensatory mechanisms directed towards normalization of relaxation. We propose that HCM is a model for diastolic heart failure and this mouse model could be valuable in studying mechanisms and treatment modalities.
肥厚型心肌病(HCM)常由编码心肌肌球蛋白结合蛋白 C(cMyBP-C)的 MYBPC3 基因突变引起。从基因突变到 HCM 表型的机制仍不完全清楚,部分原因是目前的 HCM 小鼠模型不能真实反映人类情况,并且早期肥厚会混淆对功能改变的解释。本研究的目的是评估肌球蛋白细丝 Ca(2+)敏感性和舒张功能障碍是否与 HCM 左心室肥厚(LVH)的发展相关或先于其发展。我们评估了最近开发的 Mybpc3 靶向敲入小鼠模型中,经皮和完整心肌细胞以及完整心脏的功能,该模型携带与 HCM 相关的常突变点。与野生型相比,10 周龄的纯合敲入小鼠表现出:i)经皮心室小梁肌球蛋白细丝 Ca(2+)敏感性更高,ii)完整心肌细胞舒张期肌节长度更低,Ca(2+)瞬变衰减更快,iii)超声心动图和多普勒分析显示 LVH、缩短分数降低、E/A 和 E'/A'降低以及 E/E'升高,提示收缩和舒张功能障碍。相比之下,模拟人类 HCM 情况的杂合敲入小鼠不表现出 LVH 或收缩功能障碍,但表现出更高的肌球蛋白细丝 Ca(2+)敏感性、更快的 Ca(2+)瞬变衰减和舒张功能障碍。这些数据表明,肌球蛋白细丝 Ca(2+)敏感性和舒张功能障碍是 Mybpc3 突变的早期表型后果,独立于 LVH。加速的 Ca(2+)瞬变表明存在针对松弛正常化的代偿机制。我们提出 HCM 是舒张性心力衰竭的模型,这种小鼠模型在研究机制和治疗方式方面可能具有价值。