Subbotin Vladimir M
Theor Biol Med Model. 2012 Apr 10;9:11. doi: 10.1186/1742-4682-9-11.
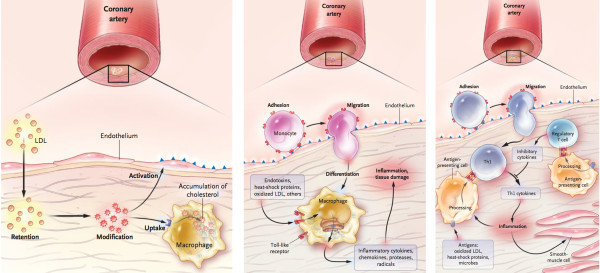
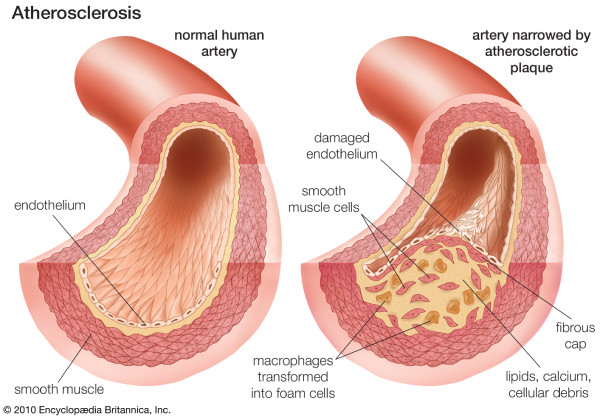
An accepted hypothesis states that coronary atherosclerosis (CA) is initiated by endothelial dysfunction due to inflammation and high levels of LDL-C, followed by deposition of lipids and macrophages from the luminal blood into the arterial intima, resulting in plaque formation. The success of statins in preventing CA promised much for extended protection and effective therapeutics. However, stalled progress in pharmaceutical treatment gives a good reason to review logical properties of the hypothesis underlining our efforts, and to reconsider whether our perception of CA is consistent with facts about the normal and diseased coronary artery.
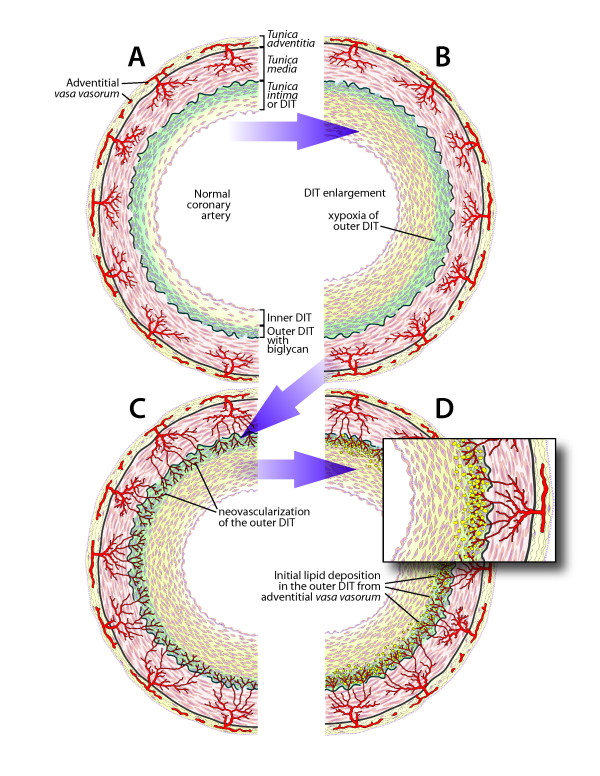
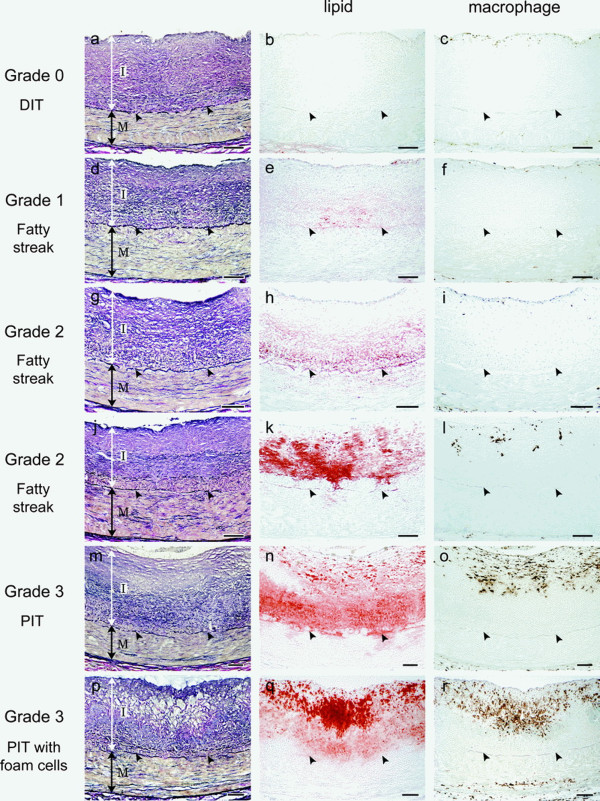
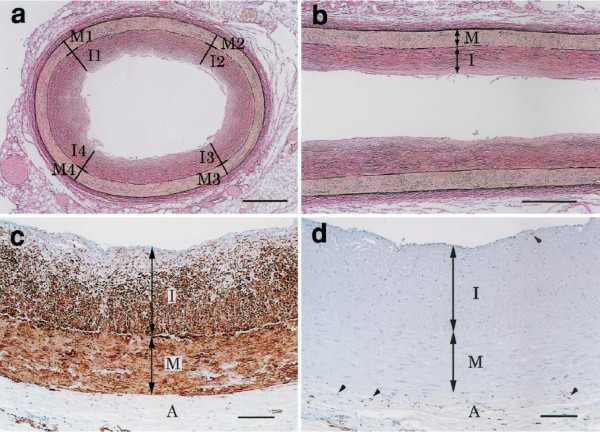
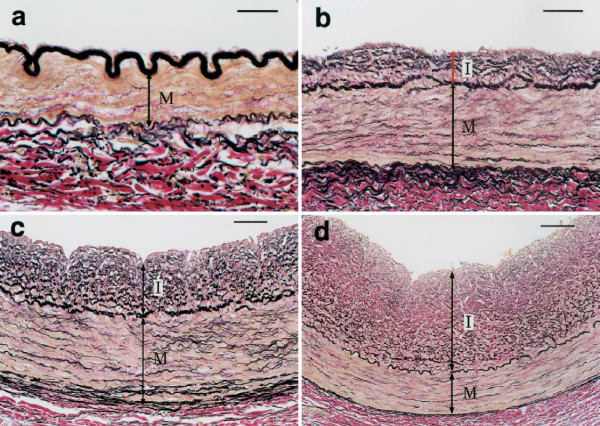
To begin with, it must be noted that the normal coronary intima is not a single-layer endothelium covering a thin acellular compartment, as claimed in most publications, but always appears as a multi-layer cellular compartment, or diffuse intimal thickening (DIT), in which cells are arranged in many layers. If low density lipoprotein cholesterol (LDL-C) invades the DIT from the coronary lumen, the initial depositions ought to be most proximal to blood, i.e. in the inner DIT. The facts show that the opposite is true, and lipids are initially deposited in the outer DIT. This contradiction is resolved by observing that the normal DIT is always avascular, receiving nutrients by diffusion from the lumen, whereas in CA the outer DIT is always neovascularized from adventitial vasa vasorum. The proteoglycan biglycan, confined to the outer DIT in both normal and diseased coronary arteries, has high binding capacity for LDL-C. However, the normal DIT is avascular and biglycan-LDL-C interactions are prevented by diffusion distance and LDL-C size (20 nm), whereas in CA, biglycan in the outer DIT can extract lipoproteins by direct contact with the blood. These facts lead to the single simplest explanation of all observations: (1) lipid deposition is initially localized in the outer DIT; (2) CA often develops at high blood LDL-C levels; (3) apparent CA can develop at lowered blood LDL-C levels. This mechanism is not unique to the coronary artery: for instance, the normally avascular cornea accumulates lipoproteins after neovascularization, resulting in lipid keratopathy.
Neovascularization of the normally avascular coronary DIT by permeable vasculature from the adventitial vasa vasorum is the cause of LDL deposition and CA. DIT enlargement, seen in early CA and aging, causes hypoxia of the outer DIT and induces neovascularization. According to this alternative proposal, coronary atherosclerosis is not related to inflammation and can occur in individuals with normal circulating levels of LDL, consistent with research findings.
一个被广泛接受的假说是,冠状动脉粥样硬化(CA)是由炎症和高水平的低密度脂蛋白胆固醇(LDL-C)导致的内皮功能障碍引发的,随后脂质和巨噬细胞从管腔血液中沉积到动脉内膜,从而形成斑块。他汀类药物在预防CA方面的成功为扩大保护范围和有效治疗带来了很大希望。然而,药物治疗进展的停滞为审视支撑我们研究工作的该假说的逻辑特性提供了充分理由,并促使我们重新思考我们对CA的认知是否与正常和病变冠状动脉的实际情况相符。
首先,必须指出的是,正常冠状动脉内膜并非如大多数出版物所声称的那样是覆盖着薄无细胞区的单层内皮,而是始终呈现为多层细胞区,即弥漫性内膜增厚(DIT),其中细胞呈多层排列。如果低密度脂蛋白胆固醇(LDL-C)从冠状动脉腔侵入DIT,那么最初的沉积物应该最靠近血液,即在DIT内层。但事实表明情况恰恰相反,脂质最初沉积在DIT外层。通过观察发现,正常的DIT始终无血管,通过从管腔扩散获取营养,而在CA中,外层DIT总是由外膜血管新生血管化。蛋白聚糖双糖链蛋白聚糖在正常和病变冠状动脉中都局限于外层DIT,对LDL-C具有高结合能力。然而,正常的DIT无血管,双糖链蛋白聚糖-LDL-C相互作用因扩散距离和LDL-C大小(20纳米)而受到阻碍,而在CA中,外层DIT中的双糖链蛋白聚糖可通过与血液直接接触提取脂蛋白。这些事实导致了对所有观察结果的最简单解释:(1)脂质沉积最初定位于外层DIT;(2)CA通常在血液LDL-C水平较高时发生;(3)在血液LDL-C水平降低时也可能出现明显的CA。这种机制并非冠状动脉所特有:例如,正常无血管的角膜在新生血管化后会积累脂蛋白,导致脂质角膜病变。
来自外膜血管的可渗透血管使正常无血管的冠状动脉DIT新生血管化是LDL沉积和CA的原因。在早期CA和衰老过程中出现的DIT增大导致外层DIT缺氧并诱导新生血管化。根据这一替代性观点,冠状动脉粥样硬化与炎症无关,可发生在LDL循环水平正常的个体中,这与研究结果一致。