Laboratory for MetaSystems Research, Quantitative Biology Center, RIKEN, Yokohama, Kanagawa, Japan.
PLoS One. 2012;7(4):e34030. doi: 10.1371/journal.pone.0034030. Epub 2012 Apr 4.
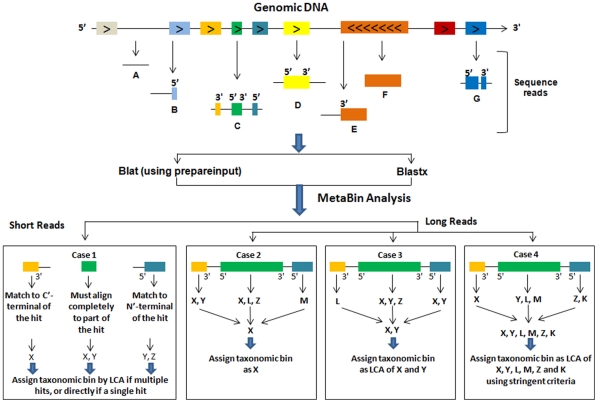
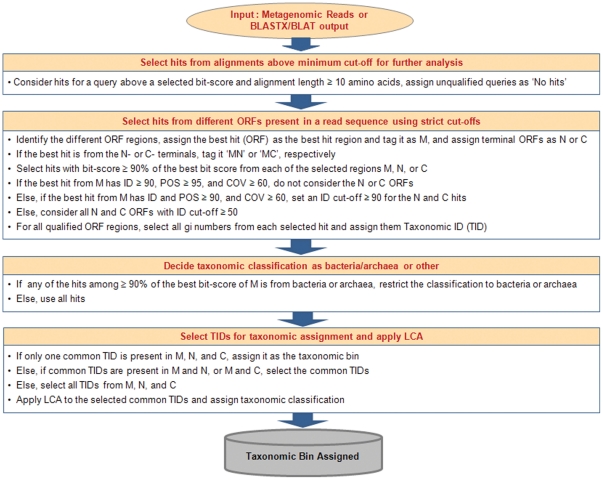
Taxonomic assignment of sequence reads is a challenging task in metagenomic data analysis, for which the present methods mainly use either composition- or homology-based approaches. Though the homology-based methods are more sensitive and accurate, they suffer primarily due to the time needed to generate the Blast alignments. We developed the MetaBin program and web server for better homology-based taxonomic assignments using an ORF-based approach. By implementing Blat as the faster alignment method in place of Blastx, the analysis time has been reduced by severalfold. It is benchmarked using both simulated and real metagenomic datasets, and can be used for both single and paired-end sequence reads of varying lengths (≥45 bp). To our knowledge, MetaBin is the only available program that can be used for the taxonomic binning of short reads (<100 bp) with high accuracy and high sensitivity using a homology-based approach. The MetaBin web server can be used to carry out the taxonomic analysis, by either submitting reads or Blastx output. It provides several options including construction of taxonomic trees, creation of a composition chart, functional analysis using COGs, and comparative analysis of multiple metagenomic datasets. MetaBin web server and a standalone version for high-throughput analysis are available freely at http://metabin.riken.jp/.
序列读取的分类分配是宏基因组数据分析中的一项具有挑战性的任务,目前的方法主要使用基于组成或同源性的方法。虽然基于同源性的方法更敏感和准确,但它们主要由于生成 Blast 比对所需的时间而受到影响。我们开发了 MetaBin 程序和网络服务器,以便使用基于 ORF 的方法进行更好的基于同源性的分类分配。通过实现 Blat 作为更快的比对方法来替代 Blastx,分析时间已经减少了几倍。它使用模拟和真实的宏基因组数据集进行了基准测试,并且可以用于不同长度(≥45 bp)的单端和双端序列读取。据我们所知,MetaBin 是唯一可用的程序,可以使用基于同源性的方法对短读取(<100 bp)进行高精度和高灵敏度的分类。MetaBin 网络服务器可以通过提交读取或 Blastx 输出来进行分类分析。它提供了几种选项,包括构建分类树、创建组成图表、使用 COGs 进行功能分析以及比较多个宏基因组数据集。MetaBin 网络服务器和用于高通量分析的独立版本可在 http://metabin.riken.jp/ 免费获得。