Center for Research in Biological Systems, University of California San Diego, La Jolla, California 92093, USA.
BMC Genomics. 2011 Sep 7;12:444. doi: 10.1186/1471-2164-12-444.
The new field of metagenomics studies microorganism communities by culture-independent sequencing. With the advances in next-generation sequencing techniques, researchers are facing tremendous challenges in metagenomic data analysis due to huge quantity and high complexity of sequence data. Analyzing large datasets is extremely time-consuming; also metagenomic annotation involves a wide range of computational tools, which are difficult to be installed and maintained by common users. The tools provided by the few available web servers are also limited and have various constraints such as login requirement, long waiting time, inability to configure pipelines etc.
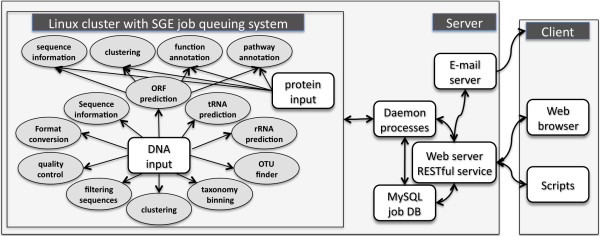
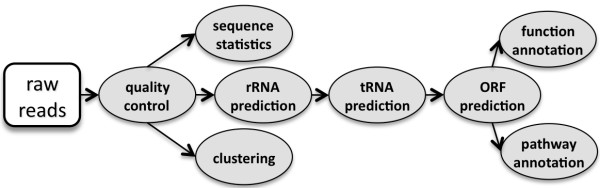
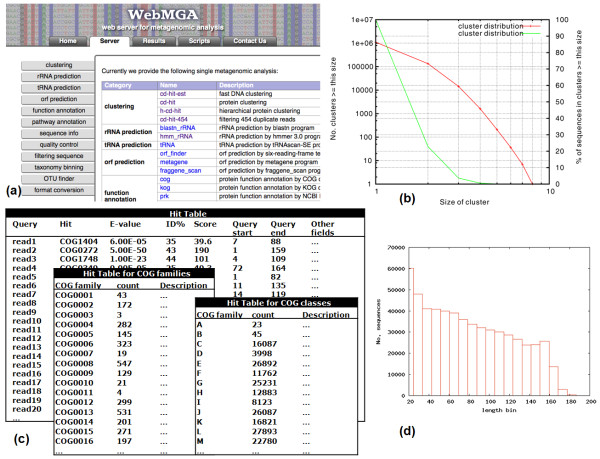
We developed WebMGA, a customizable web server for fast metagenomic analysis. WebMGA includes over 20 commonly used tools such as ORF calling, sequence clustering, quality control of raw reads, removal of sequencing artifacts and contaminations, taxonomic analysis, functional annotation etc. WebMGA provides users with rapid metagenomic data analysis using fast and effective tools, which have been implemented to run in parallel on our local computer cluster. Users can access WebMGA through web browsers or programming scripts to perform individual analysis or to configure and run customized pipelines. WebMGA is freely available at http://weizhongli-lab.org/metagenomic-analysis.
WebMGA offers to researchers many fast and unique tools and great flexibility for complex metagenomic data analysis.
宏基因组学通过非培养测序来研究微生物群落。随着下一代测序技术的进步,由于序列数据的数量巨大且复杂性高,研究人员在宏基因组数据分析方面面临着巨大的挑战。分析大型数据集非常耗时;此外,宏基因组注释涉及广泛的计算工具,普通用户很难安装和维护这些工具。少数可用的网络服务器提供的工具也受到限制,存在登录要求、长时间等待、无法配置管道等各种限制。
我们开发了 WebMGA,这是一个用于快速宏基因组分析的可定制的网络服务器。WebMGA 包括 20 多种常用工具,如 ORF 调用、序列聚类、原始读数的质量控制、测序伪影和污染的去除、分类分析、功能注释等。WebMGA 为用户提供了快速的宏基因组数据分析,使用快速有效的工具,这些工具已经在我们的本地计算机集群上实现了并行运行。用户可以通过网络浏览器或编程脚本访问 WebMGA,以执行单个分析或配置和运行自定义管道。WebMGA 可免费在 http://weizhongli-lab.org/metagenomic-analysis 获得。
WebMGA 为研究人员提供了许多快速而独特的工具,以及用于复杂宏基因组数据分析的极大灵活性。