Institute of Infection, Immunity and Inflammation, College of Medical, Veterinary and Life Sciences, University of Glasgow, Sir Graeme Davies Building, Glasgow G12 8QQ, UK.
BMC Bioinformatics. 2012 Apr 27;13:63. doi: 10.1186/1471-2105-13-63.
Outer membrane proteins (OMPs) of Pasteurella multocida have various functions related to virulence and pathogenesis and represent important targets for vaccine development. Various bioinformatic algorithms can predict outer membrane localization and discriminate OMPs by structure or function. The designation of a confident prediction framework by integrating different predictors followed by consensus prediction, results integration and manual confirmation will improve the prediction of the outer membrane proteome.
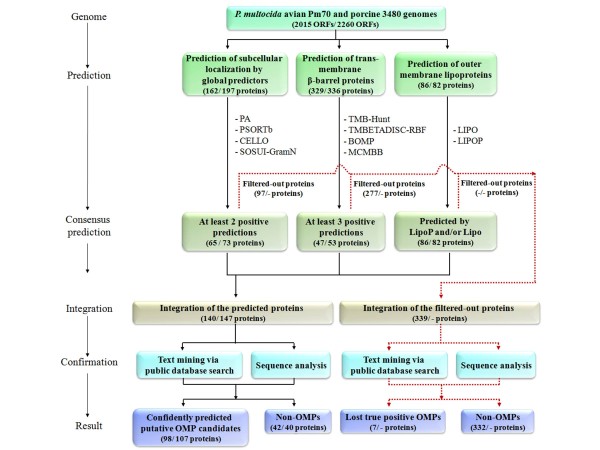
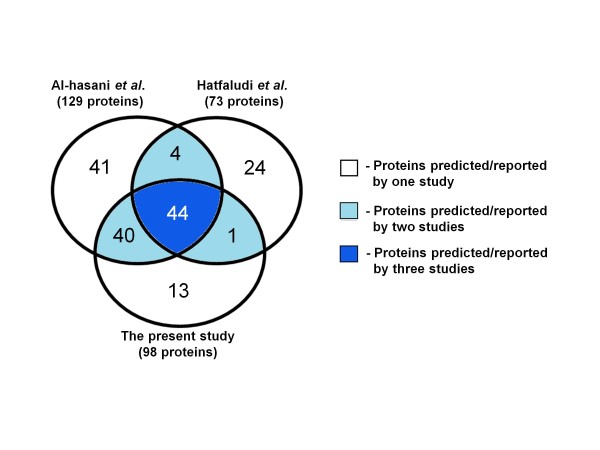
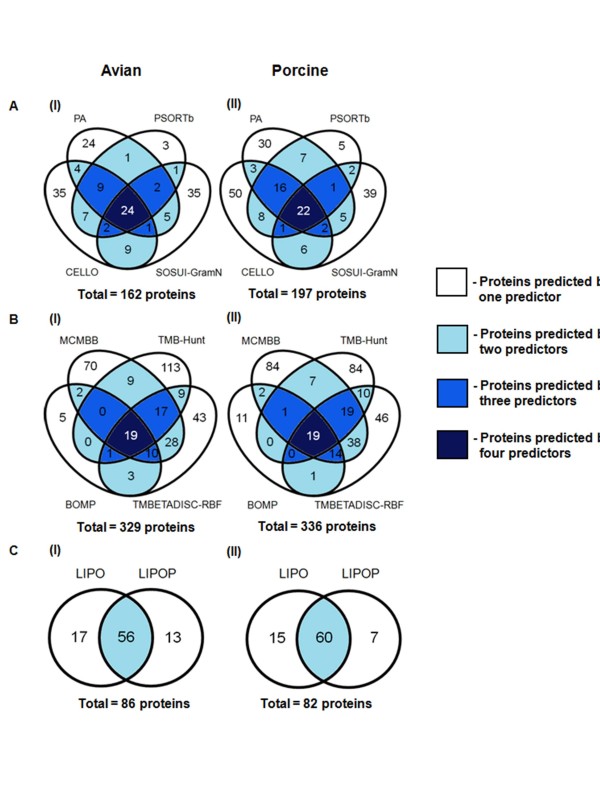
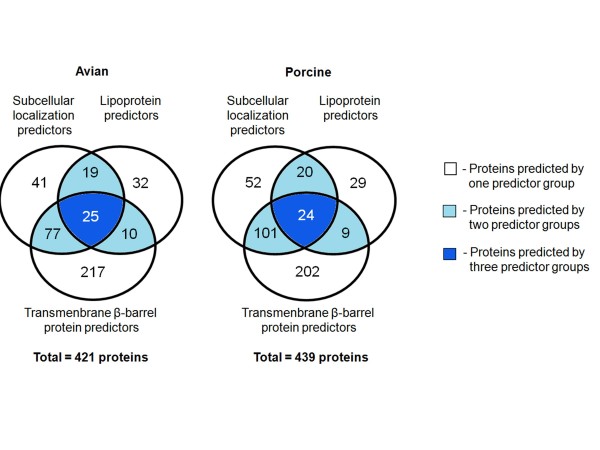
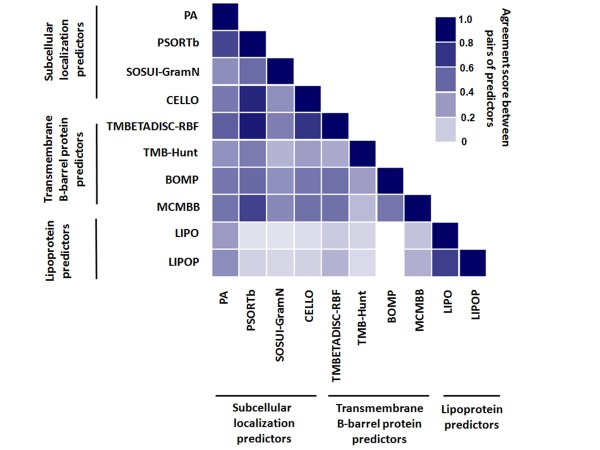
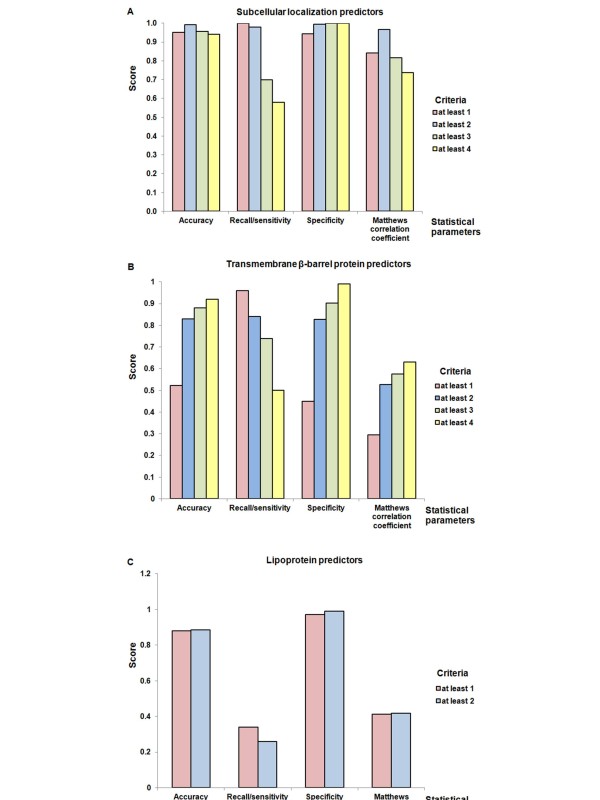
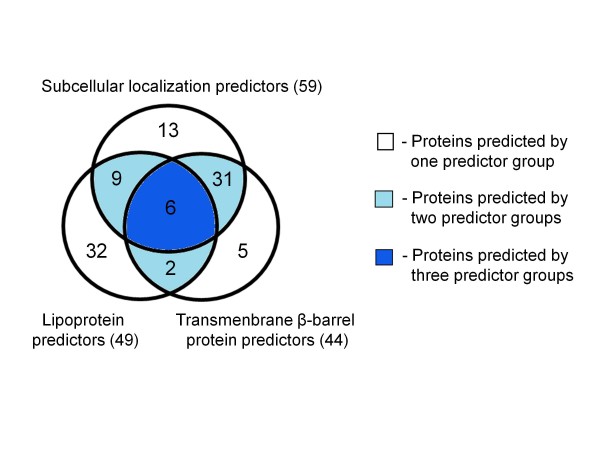
In the present study, we used 10 different predictors classified into three groups (subcellular localization, transmembrane β-barrel protein and lipoprotein predictors) to identify putative OMPs from two available P. multocida genomes: those of avian strain Pm70 and porcine non-toxigenic strain 3480. Predicted proteins in each group were filtered by optimized criteria for consensus prediction: at least two positive predictions for the subcellular localization predictors, three for the transmembrane β-barrel protein predictors and one for the lipoprotein predictors. The consensus predicted proteins were integrated from each group into a single list of proteins. We further incorporated a manual confirmation step including a public database search against PubMed and sequence analyses, e.g. sequence and structural homology, conserved motifs/domains, functional prediction, and protein-protein interactions to enhance the confidence of prediction. As a result, we were able to confidently predict 98 putative OMPs from the avian strain genome and 107 OMPs from the porcine strain genome with 83% overlap between the two genomes.
The bioinformatic framework developed in this study has increased the number of putative OMPs identified in P. multocida and allowed these OMPs to be identified with a higher degree of confidence. Our approach can be applied to investigate the outer membrane proteomes of other Gram-negative bacteria.
多杀巴斯德氏菌的外膜蛋白(OMPs)具有与毒力和发病机制相关的各种功能,是疫苗开发的重要靶标。各种生物信息学算法可以预测外膜定位,并通过结构或功能来区分 OMPs。通过整合不同的预测器进行有信心的预测框架的指定,然后进行共识预测、结果整合和手动确认,将提高外膜蛋白质组预测的准确性。
在本研究中,我们使用了 10 种不同的预测器,分为三个组(亚细胞定位、跨膜β-桶蛋白和脂蛋白预测器),从两种可用的多杀巴斯德氏菌基因组(禽源株 Pm70 和非致毒性猪源株 3480)中鉴定潜在的 OMPs。每个组的预测蛋白都经过优化的共识预测标准进行过滤:亚细胞定位预测器至少有两个阳性预测,跨膜β-桶蛋白预测器有三个,脂蛋白预测器有一个。共识预测的蛋白从每个组整合到一个单一的蛋白列表中。我们进一步纳入了一个手动确认步骤,包括针对 PubMed 的公共数据库搜索和序列分析,例如序列和结构同源性、保守基序/结构域、功能预测和蛋白质-蛋白质相互作用,以提高预测的置信度。结果,我们能够从禽源株基因组中自信地预测 98 个潜在的 OMPs,从猪源株基因组中预测 107 个 OMPs,两个基因组之间有 83%的重叠。
本研究中开发的生物信息学框架增加了多杀巴斯德氏菌中潜在 OMPs 的数量,并提高了这些 OMPs 的鉴定置信度。我们的方法可以应用于研究其他革兰氏阴性菌的外膜蛋白质组。