Lu Shih-Jen, Chong Fok-Ching
Department of Electrical Engineering, National Taiwan University, Taipei 10617, Taiwan.
Department of Research and Development, BroadMaster Biotech Co., Ltd.: 7F., No.168-2, Liancheng Rd., Zhonghe Dist., New Taipei City 23553, Taiwan.
Int J Mol Sci. 2012;13(4):4496-4507. doi: 10.3390/ijms13044496. Epub 2012 Apr 10.
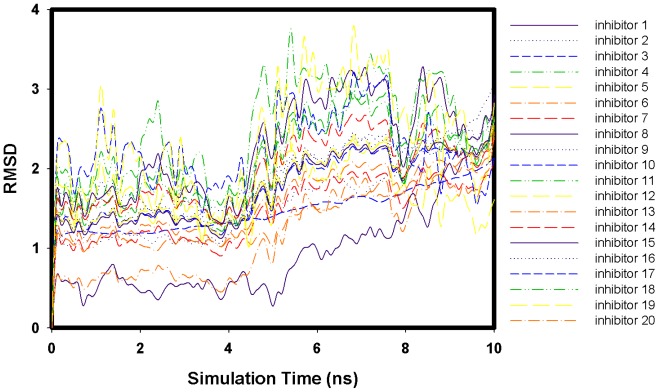
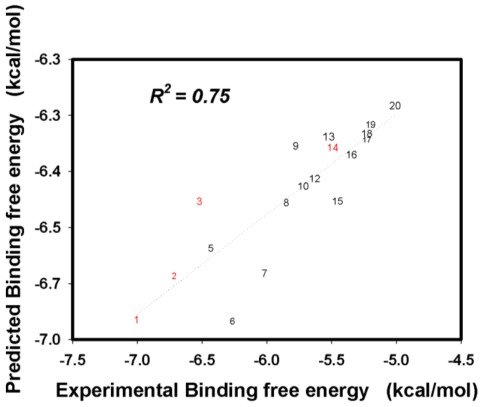
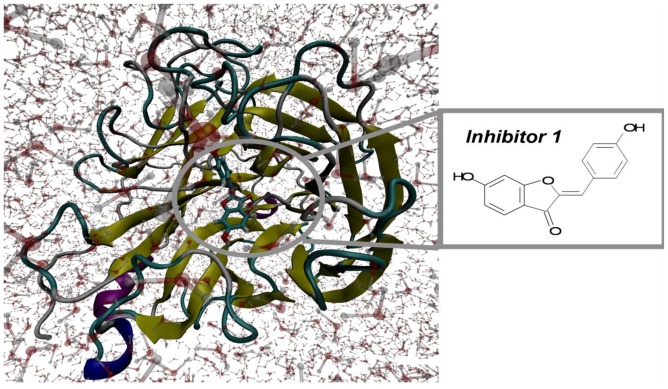
Control of flavonoid derivatives inhibitors release through the inhibition of neuraminidase has been identified as a potential target for the treatment of H1N1 influenza disease. We have employed molecular dynamics simulation techniques to optimize the 2009 H1N1 influenza neuraminidase X-ray crystal structure. Molecular docking of the compounds revealed the possible binding mode. Our molecular dynamics simulations combined with the solvated interaction energies technique was applied to predict the docking models of the inhibitors in the binding pocket of the H1N1 influenza neuraminidase. In the simulations, the correlation of the predicted and experimental binding free energies of all 20 flavonoid derivatives inhibitors is satisfactory, as indicated by R(2) = 0.75.
通过抑制神经氨酸酶来控制类黄酮衍生物抑制剂的释放已被确定为治疗H1N1流感疾病的一个潜在靶点。我们采用分子动力学模拟技术对2009年H1N1流感神经氨酸酶的X射线晶体结构进行了优化。化合物的分子对接揭示了可能的结合模式。我们将分子动力学模拟与溶剂化相互作用能技术相结合,用于预测抑制剂在H1N1流感神经氨酸酶结合口袋中的对接模型。在模拟中,所有20种类黄酮衍生物抑制剂的预测结合自由能与实验结合自由能之间的相关性令人满意,R(2) = 0.75表明了这一点。