School of Pharmacy, Centre for Biomolecular Sciences, University of Nottingham, University Park, Nottingham, Nottinghamshire NG7 2RD, UK.
ChemMedChem. 2012 Aug;7(8):1435-46. doi: 10.1002/cmdc.201200107. Epub 2012 May 29.
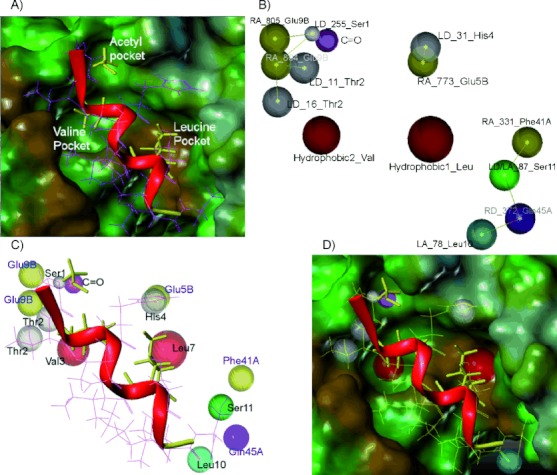
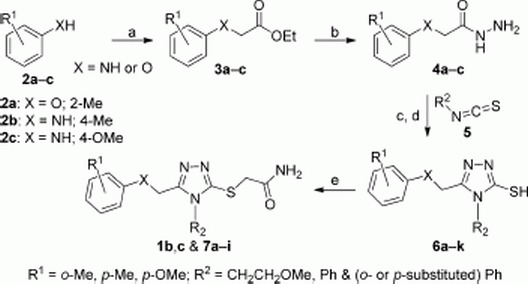
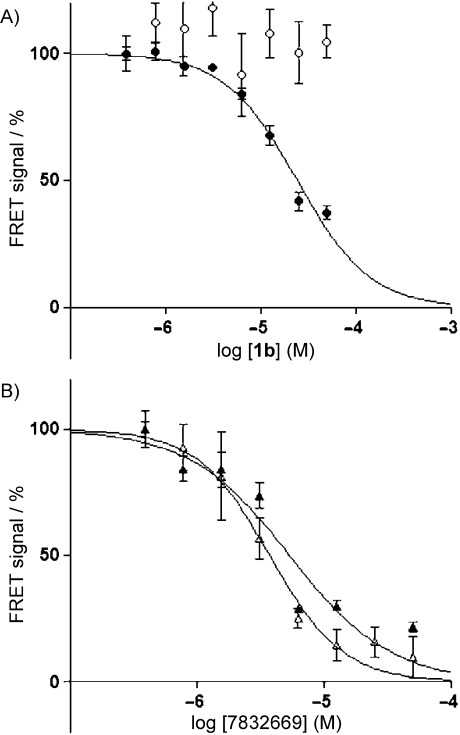
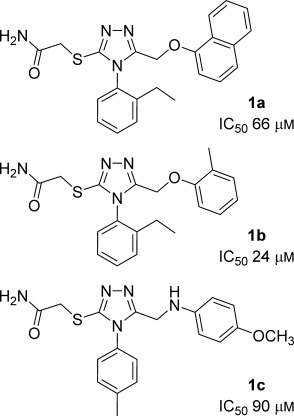
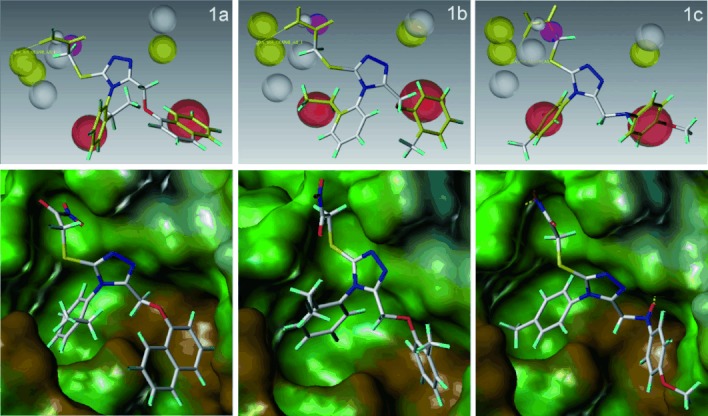
Protein interactions are increasingly appreciated as targets in small-molecule drug discovery. The interaction between the adapter protein S100A10 and its binding partner annexin A2 is a potentially important drug target. To obtain small-molecule starting points for inhibitors of this interaction, a three-dimensional pharmacophore model was constructed from the X-ray crystal structure of the complex between S100A10 and annexin A2. The pharmacophore model represents the favourable hydrophobic and hydrogen bond interactions between the two partners, as well as spatial and receptor site constraints (excluded volume spheres). Using this pharmacophore model, UNITY flex searches were carried out on a 3D library of 0.7 million commercially available compounds. This resulted in 568 hit compounds. Subsequently, GOLD docking studies were performed on these hits, and a set of 190 compounds were purchased and tested biochemically for inhibition of the protein interaction. Three compounds of similar chemical structure were identified as genuine inhibitors of the binding of annexin A2 to S100A10. The binding modes predicted by GOLD were in good agreement with their UNITY-generated conformations. We synthesised a series of analogues revealing areas critical for binding. Thus computational predictions and biochemical screening can be used successfully to derive novel chemical classes of protein-protein interaction blockers.
蛋白质相互作用作为小分子药物发现的靶点越来越受到重视。衔接蛋白 S100A10 与其结合伴侣膜联蛋白 A2 之间的相互作用是一个潜在的重要药物靶点。为了获得该相互作用抑制剂的小分子起始点,根据 S100A10 与膜联蛋白 A2 复合物的 X 射线晶体结构构建了三维药效团模型。药效团模型代表了两个配体之间有利的疏水和氢键相互作用,以及空间和受体部位限制(排除体积球)。使用该药效团模型,在 3D 库中进行了 0.7 百万种商业可得化合物的 UNITY flex 搜索。这产生了 568 个命中化合物。随后,对这些命中化合物进行了 GOLD 对接研究,并购买了一组 190 种化合物进行生化测试,以抑制蛋白质相互作用。三种具有相似化学结构的化合物被鉴定为膜联蛋白 A2 与 S100A10 结合的真正抑制剂。GOLD 预测的结合模式与其 UNITY 生成的构象非常吻合。我们合成了一系列类似物,揭示了结合的关键区域。因此,计算预测和生化筛选可成功用于推导新型蛋白质-蛋白质相互作用阻滞剂的化学类别。